
基因變異普遍存在於同一物種內的不同個體中,如人與人之間的基因組是不完全一樣的(即是多態的),彼此之間都存在着一些差異,即使是和父母或是兄弟姐妹之間去比較。這種差異也是基因組多態性的來源,通過外在和內在特徵表現出來,比如頭髮和眼睛顏色,高矮胖瘦,抵抗力等。這些差異也是造成我們彼此之間不同的一個重要原因。
基因變異是一個相對的概念,只有在彼此的比較中才有存在意義。基因變異主要分為三大類:
- 單核苷酸變異,通常稱為單核苷酸多態性(single nucleotide polymorphism),就是單個DNA鹼基的不同,簡稱SNP;
- 小的Indel(Insertion 和 Deletion的簡),指的是在基因組的某個位置上所發生的小片段序列的插入或者刪除,長度通常在50 bp以下;
- 大的結構性變異,這種類型比較多,包括長度在50 bp以上的長片段序列的插入或者刪除、染色體倒位,染色體內部或染色體之間的序列易位,拷貝數變異,以及一些形式更為複雜的變異。
為了和SNP變異作區分,第2和第3類變異通常也被稱為基因組結構性變異(Structural variation,簡稱SV)。
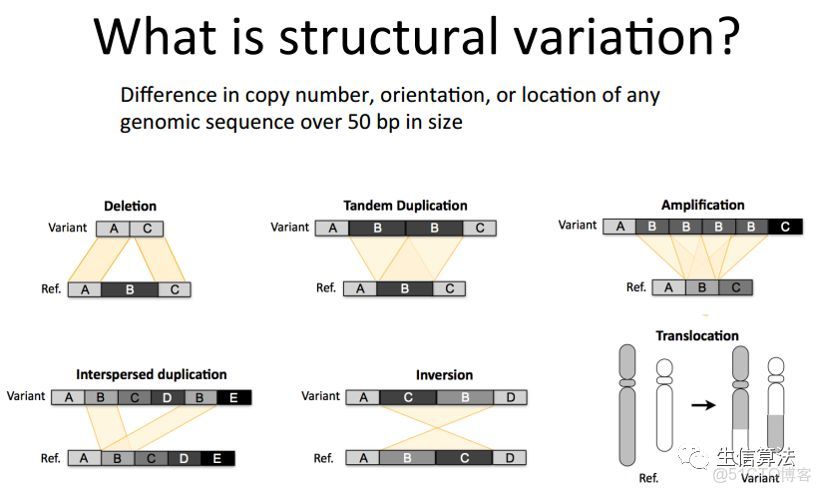
結構變異長度少則幾十bp,多則幾萬bp,對其檢測帶來了極大挑戰。研究人員發現SV對基因組的影響比起SNP來説還要大,而且基因組上的SVs比起SNP而言,似乎更能用於解釋人類羣體多樣性的特徵。再者,一些SVs往往和一些疾病(包括癌症、自閉症、老年痴呆症)的發生相關聯,甚至還是其致病的誘因。因此對SVs進行檢測具有重要的意義。
二代測序技術產生的序列雖然精度高,但讀段短(100~500 bp),不能跨越大多數長度較長的SVs區域,但基於單分子測序的三代測序技術(如PacBio,牛津納米孔測序)可以測得長達100 kpb的序列,輕鬆跨越各種SVs區域,為SVs檢測帶來方便。但其錯誤率較高(~15%),因此需要專門的SVs檢測算法。PBHoney是較早利用三代序列進行SVs檢測的軟件。
PBHoney方法
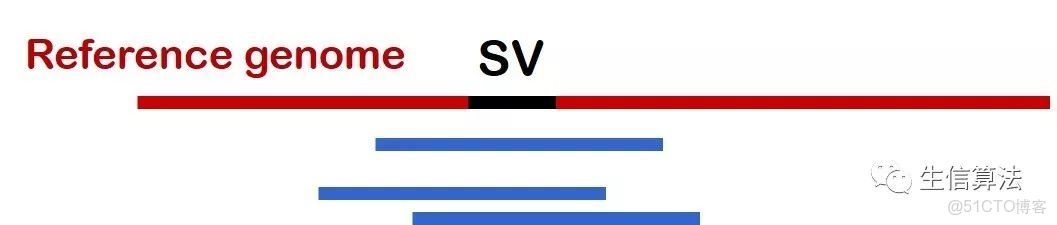
PBHoney主要利用兩種序列比對情況進行檢測。如下圖所示,一種是Interrupted long-read mapping,一種是 Intra-read discordance。主要原因在於對於一條比對的三代序列,比對情況可以分為這兩種。
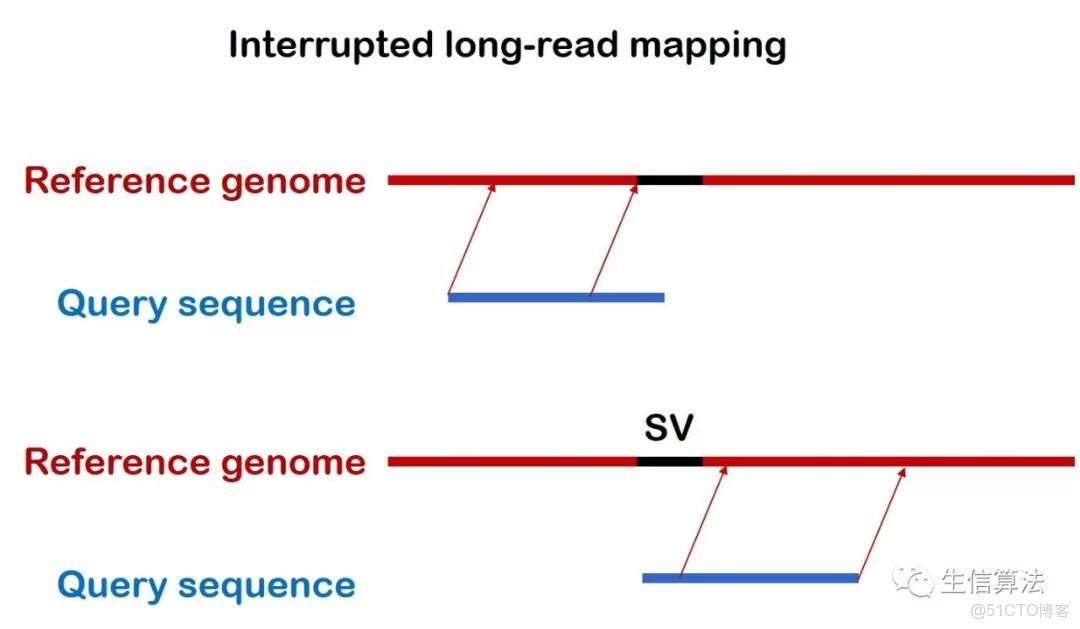
Interrupted long-read mapping 比對示意圖
所謂Interrupted long-read mapping ,就是這條序列只比對了一部分,其前綴或者後綴的序列片段沒有比對上去。
下圖是 Intra-read discordance比對示意圖,所謂 Intra-read discordance,就是整條序列都比對到參考基因組上,但某些比對區域由於SVs存在比對質量較低,存在較多非匹配狀態。
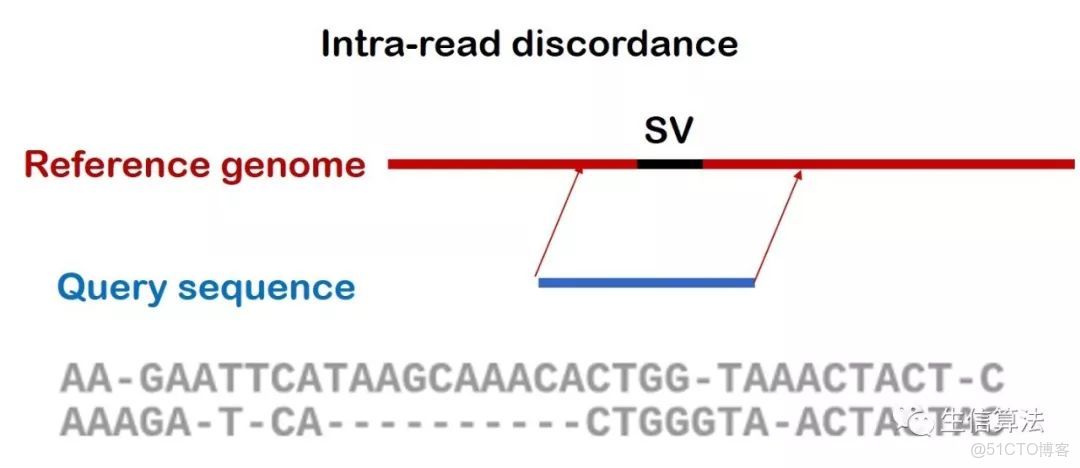
Intra-read discordance 比對示意圖
PBHoney首先採用三代序列比對算法BLASR進行序列比對,然後針對這兩種比對情況做相應的處理,找出SVs區域。
處理 Interrupted long-read mapping序列
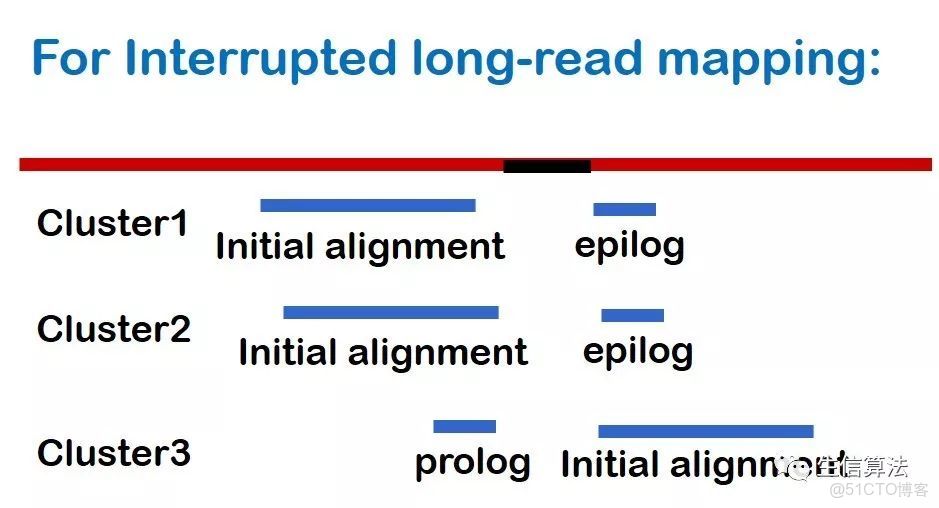
首先將提取沒有比對的前綴或者後綴,如果長度大於200 bp,再將這些片段通過BLASR比對到參考基因組上。然後與原來比對的序列(initial alignment)組成piece-alignment,如下圖所示。後綴比對的片段稱為epilog,前綴比對的片段稱為prolog。
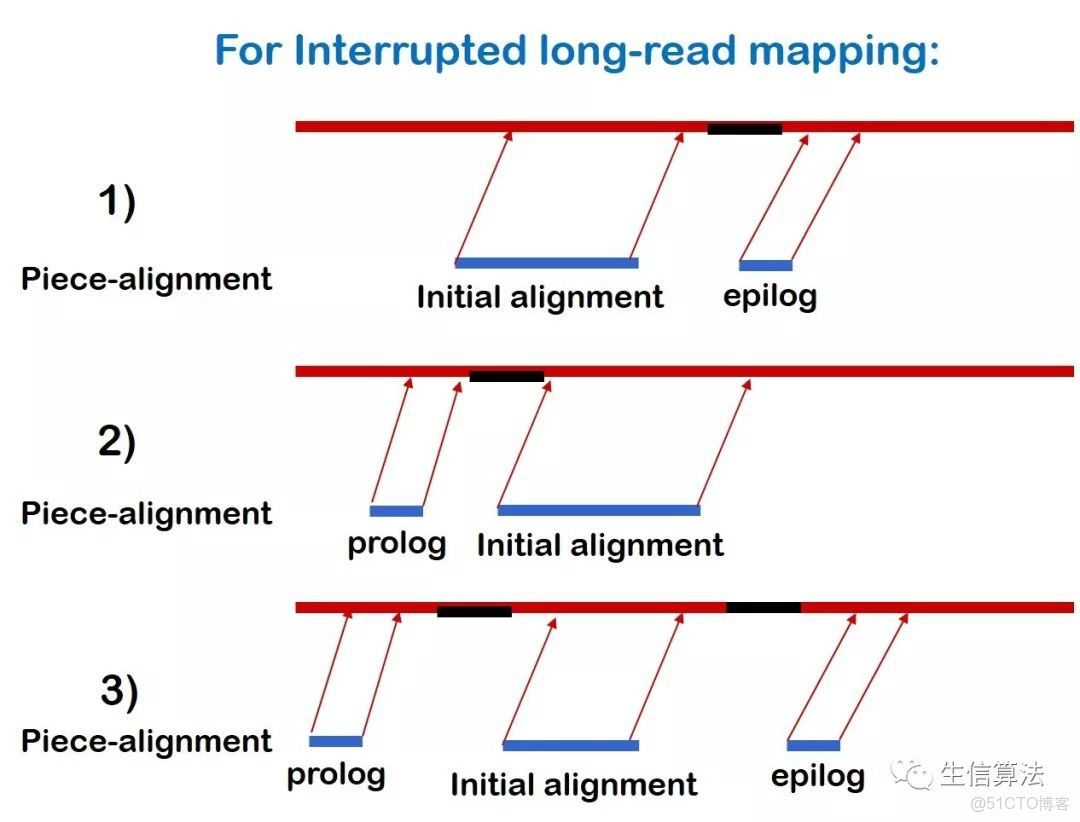
然後對每個piece-alignment進行聚類處理:
- 只考慮包含initial alignment 和 epilog (或prolog)的piece-alignment。如果epilog和prolog都存在,(如上圖3)所示,只考慮比對質量高的。
- 保留initial alignment 和 epilog (或prolog)之間的距離小於200 bp的piece-alignment。200 bp是默認值,用户可自己設置。保留下來的piece-alignment稱為一個cluster。
- SVs的位置(起始位置和結束位置)就是每個cluster中比對位置的平均值。如下圖所示。
處理 Intra-read discordance 序列
對於完整比對上的序列,如上圖所示,通過以下步驟進行處理:
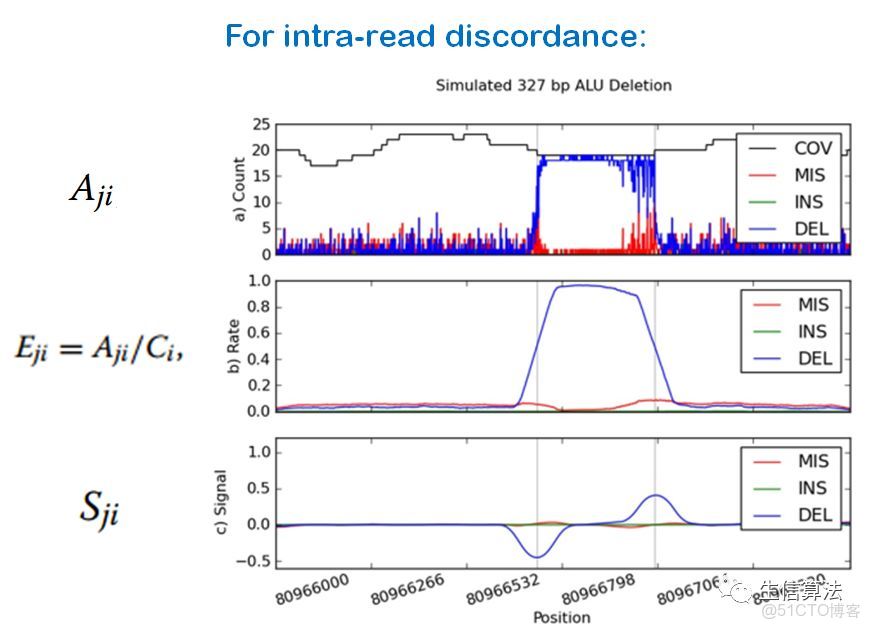
1. 計算比對到基因組序列每個位置的替換(mismatch)、插入、刪除鹼基個數、及覆蓋率。構成一個4×G大小的數組A,其中G是參考基因組的序列長度。
2. 然後計算基因組每個位置的錯誤率大小:

其中j=1,2,3,4. Ci表示覆蓋率。
3. 通過下面公式對E矩陣進行變換:
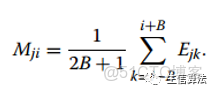
4. 再次對M矩陣進行變換:
 .")
其中步驟3)和4)目的是尋找一些差異比較大的值,更好判斷是否是SV。
下圖是通過以上步驟檢測一個刪除SV的示意圖。可以看出步驟3)和4)可以將A柱狀圖轉變成脈衝信號,方便SV檢測。
結果
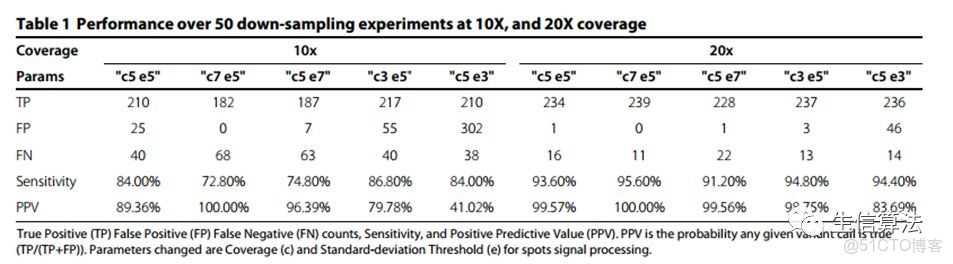
下表是PBHoney的檢測結果,可以看出其靈敏度和PPV值還是不錯的,這篇文章是以軟件類(software)形式發表的,沒有和別的方法進行比較。
總結
SVs檢測對於疾病治療具有重要意義,隨着三代測序技術的不斷髮展,其測序準確性和序列長度會不斷改善,相信基於三代序列的結構變異檢測會得到越來越多的關注。
參考文獻
- English A C, Salerno W J, Reid J G. PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC bioinformatics, 2014, 15(1): 180.
-