
文獻數據庫:https://pubmed.ncbi.nlm.nih.gov/
Filters applied: from 2025/4/19 - 11/25
主要列出文章目錄,個別文獻含圖表。文章思路解讀,後續逐步更新
1. Hypohidrotic ectodermal dysplasia: association between EDA mutations and hypotrichosis - a case series
少汗性外胚層發育不良:EDA突變與少毛症的關聯—病例系列研究
作者: Li Y, Zeng Q, Cao QY, Zhao AQ, Li M, Zhu NW。復旦大學附屬兒童醫院皮膚科
Eur J Dermatol. 《歐洲皮膚病學雜誌》
影響因子:1.6;中科院分區:4區。投稿難度:相對較低,審稿速度較慢,版面費較低
DOI: 10.1684/ejd.2025.4949
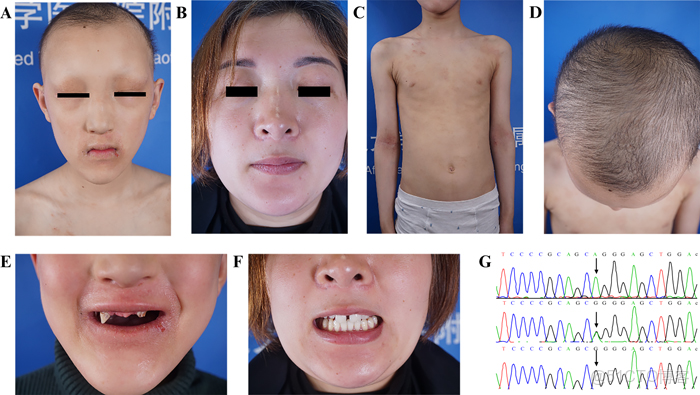
圖1. A) 眉毛和睫毛缺失,伴有眶周色素沉着。B) 先證者母親的面部特徵正常。C) 皮膚乾燥和乳腺發育不全。D) 少毛症。E) 少齒、圓錐牙。F) 先證者母親的牙齒錯位、圓錐牙。G) 先證者、母親和父親的Sanger測序峯圖 (從上到下) 顯示 EDA 基因外顯子7中存在錯義突變 (c.871G>a,p.G291A) 。
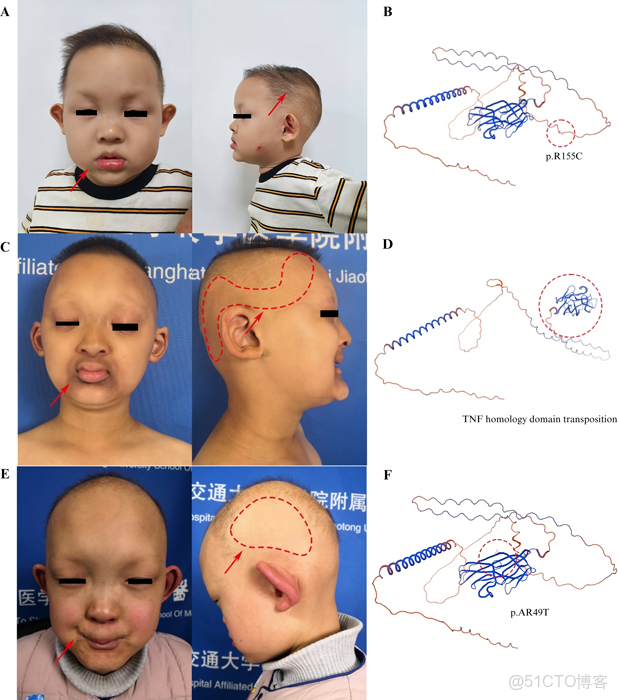
圖3. A) 毛髮減少,但頭皮毛髮能完整覆蓋。B) 先證者19的 EDA 蛋白的空間構象建模。C) 毛髮減少、局灶性脱髮、口周色素沉着。D) 先證者10的 EDA 蛋白的空間構象建模。E) 脱髮,局灶性脱髮。F) 先證者13 EDA 蛋白的空間構象建模。
2. Compound genetic burden in oculo-facio-cardio-dental (OFCD) syndrome: surgical risk stratification with co-occurring BCOR and MYLK mutations
眼-面-心-牙綜合徵的複合遺傳負擔:BCOR與MYLK共突變的手術風險分層
作者: Kang S, Perez AM, Smith C, Chang TC, Bademci G
Ophthalmic Genet. 影響因子:1.4;中科院分區:4區
DOI: 10.1080/13816810.2025.2582609
3. Genomic Confluence: When Cerebrotendinous Xanthomatosis, Klinefelter Syndrome, and a BRCA2 Variant Intersect
基因組交匯:當腦腱黃瘤病、克氏綜合徵與BRCA2變異相遇
作者: Pachajoa H, Bonilla S, Nieva-Posso DA
Int J Mol Sci. 影響因子:4.9;中科院分區:生物學2區;JCR分區:Q1
DOI: 10.3390/ijms262110510
多位點致病性變異 — 當多種遺傳疾病共存於單個個體中時,這種情況很少見,但在基因組醫學時代越來越被人們所認識。報告此類病例對於提高診斷準確性、完善臨牀管理和遺傳諮詢至關重要。我們描述了1例具有複雜表型的兒科病例,含兩種不同的遺傳診斷結果,分別是:腦脊髓炎性黃瘤病 (CTX),一種罕見的常染色體隱性脂質儲存障礙,由 CYP27A1 基因的雙等位基因突變引起,以及Klinefelter綜合徵。患者出生於近親家庭,表現為神經系統症狀、胃腸功能障礙、內分泌異常和畸形特徵。此外,在BRCA2中偶然檢測到一種雜合致病性變異,與遺傳性癌症易感性相關。

4. Genetic variants in HELB contribute to premature ovarian insufficiency and early age of natural menopause
HELB基因變異導致早發性卵巢功能不全和自然絕經年齡提前
作者: Pan Y, Yu Y, Mo J, Ren S, Zhou Z, Yang X, Liu Y, Zhang F, You Y, Zhang X, Wu Y。復旦大學生命科學學院,人類表型組研究所;南昌大學江西醫學院附屬第二醫院,江西醫學科學院生物醫學創新研究所;中國人民解放軍總醫院婦產科;復旦大學國家實驗生物學教育示範中心等
JCI Insight. 影響因子:6.3;中科院分區:1區;JCR分區:Q1
DOI: 10.1172/jci.insight.191122
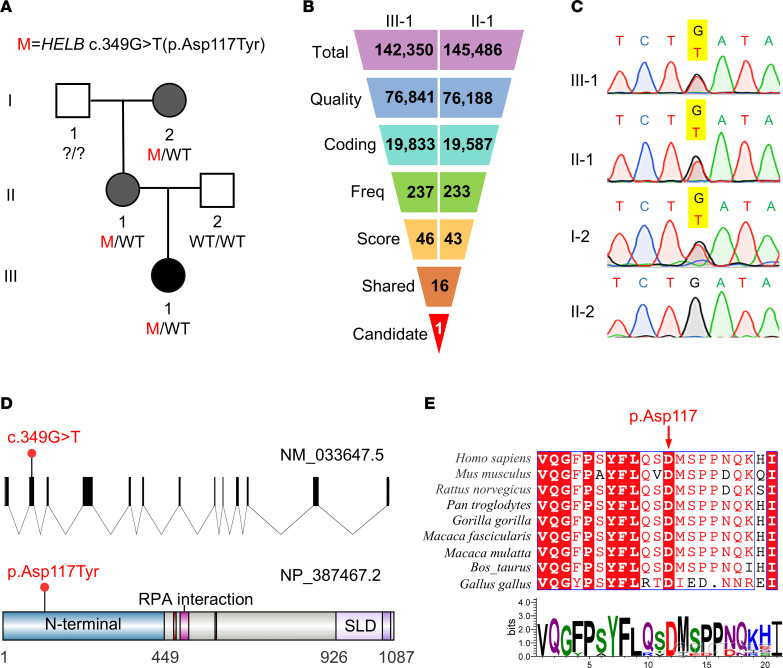
圖1. 在一個患有POI (卵巢早衰)和早發絕經的中國家庭中,鑑定 HELB 基因的有害變異。(A) 在一個患有POI和絕經早期的中國家庭中發現了HELB的雜合錯義變異 c.349G>T (p.Asp117Tyr)。黑色填充符號表示患有POI的先證者,灰色填充符號表示受早期更年期影響的家庭成員。(B) 基於質量、頻率、有害性和生物學意義的WES過濾策略的流程圖。篩選條件包括質量 (具有高/中等基因型質量的變異:測序深度>30,基因型質量>30,無低質量基因型);編碼 (外顯子或剪接位點變體);頻率 (千人基因組計劃項目[1000g]、gnomAD和ExAC數據庫中的次要等位基因頻率<0.001);評分 (SIFT、PolyPhen-2、MutationTaster和CADD_phred預測具有破壞性的變異>25);並且共享 (III-1 和 II-1 共享的變異)。(C) 受影響個體 HELB 變異的Sanger測序驗證。突變位置以黃色突出顯示。(D) 人類基因 HELB(NM_033647.5) 和人類HELB蛋白 (NP_387467.2)的示意圖。突變位置用紅點表示。 (E) 不同物種中 HELB 氨基酸序列的保守性分析。紅色箭頭表示受已鑑定的變異所影響的 Asp117 殘基。
5. Identification of a VHL germline deletion in a family with Von Hippel-Lindau syndrome using MLPA-NGS
使用MLPA-NGS鑑定 Von Hippel-Lindau 綜合徵家系中的VHL種系缺失
作者: Yang Y, Ren X, Xia C, Zhang Y, Song X, Tang X, Du C, Xu W, Weng W
BMC Med Genomics. DOI: 10.1186/s12920-025-02252-y
Von Hippel-Lindau病是一種罕見的遺傳性神經皮膚疾病,特徵是多器官的良性或惡性腫瘤。發病率為1/36,000,為具有可變外顯率的常染色體顯性遺傳。
6. A novel HPS3 pathogenic nonsense variant associated with Hermansky-Pudlak syndrome type 3 and a platelet dysfunction
新型HPS3致病性無義變異與Hermansky-Pudlak綜合徵3型及血小板功能障礙相關
作者: Chouman C, Salhab S, Martella S, Mousawi Z, Assi A, Chebly A, El Shamieh S
Mol Biol Rep. DOI: 10.1007/s11033-025-11142-6
7. Excessive Glycine Loop Variations in the Keratin 10 Tail Domain and Implications for Skin Fragility
Keratin 10尾部結構域中甘氨酸環過度變異及其對皮膚脆性的影響
作者: Mi Z, Yu Y, Wang Z, Bai F, Yu G, Li B, Dai Y, Lin Y, Sang X, Han J, Tu X, Wang Y, Cao N, Liu H, Zhang F
Br J Dermatol. DOI: 10.1093/bjd/ljaf436
8. Complex de novo structural variants are an underestimated cause of rare disorders
複雜的新發結構變異是罕見病的被低估的原因
作者: Jung H, Yang TP, Walker S, Danecek P, Garcia-Salinas OI, Neville MDC, Christopher J, Cortés-Ciriano I, Firth H, Scally A, Hurles M, Campbell P, Rahbari R
Nat Commun. DOI: 10.1038/s41467-025-64722-2
9. Using Whole Exome Sequencing to Identify Genetic Causes of Neurodevelopmental Disorders in a Cohort of 11 Patients: A Single Center Experience
使用全外顯子組測序鑑定11例神經發育障礙患者的遺傳病因:單中心經驗
作者: Tompa M, Sinko G, Mally J, Karteszi J, Kalman B
Int J Mol Sci. DOI: 10.3390/ijms262010176
10. Detailed assessment of rare and common TERT variation in a family with a telomere biology disorder
對一例端粒生物學疾病家系中罕見和常見TERT變異的詳細評估
作者: Zeigler LP, Florez-Vargas O, Altintas B, Niewisch MR, Zhou W, Giri N, Rafati M, Poeschla M, Sankaran VG, Lai TP, Aviv A, Jones K, Luo W, Liu J, McReynolds LJ, Zhao T, Prokunina-Olsson L, Savage SA
HGG Adv. DOI: 10.1016/j.xhgg.2025.100536
11. Deciphering ALS-linked genetic variants in indian patients using targeted and exome sequencing approaches
使用靶向測序和外顯子組測序方法解讀印度患者的ALS (漸凍症)相關遺傳變異
作者: Reza S, Handique J, Sharma P, Mathew S, Bari S, Tyagi N, Sharma C, Panda S, Chowdhury D, Laskar S, Shaji CV, Joshi D, Kp D, Cherian A, Desai S, Gourie Devi M, Srivastava AK, Faruq M
DOI: 10.1080/21678421.2025.2574681
ALS:Amyotrophic lateral sclerosis 肌萎縮側索硬化症,即漸凍症。
12. Genomics of pregnancy loss
妊娠丟失的基因組學
作者: Nikitina TV, Fonova EA, Lebedev IN
DOI: 10.1007/s10815-025-03726-9
妊娠丟失 pregnancy loss 即反覆流產。反覆妊娠丟失(RPL) 是指 連續發生2次或更多次妊娠失敗 (包括自然流產、胎停育等),是育齡女性常見的生育問題。 其病因複雜,可能與染色體異常、子宮結構異常、內分泌紊亂、免疫因素、血栓傾向等有關。
13. Case Report: Dual molecular diagnosis of gain-of-function STAT1 mutation and regulatory STAT3 variant in a patient with a hyper-IgE-like phenotype
病例報告:一名高IgE樣表型患者的功能獲得性STAT1突變和調控性STAT3變異的雙重分子診斷
作者: Yaakoubi R, Mekki N, Ben Chehida A, Benhammadi A, Chan KW, Leung D, Gharsallah C, Guerfali FZ, Barbouche MR, Lau YL, Ben-Ali M, Ben-Mustapha I
DOI: 10.3389/fimmu.2025.1646761
14. Exome sequencing reveals new insights into the germline landscape of inflammatory breast cancer among Tunisian patients
外顯子組測序揭示突尼斯患者炎性乳腺癌的胚系(變異)圖景新見解
作者: Boujemaa M, Hamdi Y, Guidara S, Souissi A, Bouaziz H, Mejri N, Aloulou S, Mahjoub N, Chaabane K, Hakim H, Boussen H, Dhiab TB, Kamoun S, Ayadi R, Driss M, Ayadi A, Radouani F, Fakhkhari M, Khyatti M, Abdelhak S, Sadki K, Boubaker MS, Rebai A, Cherif B
J Transl Med. 影響因子:7.5;中科院分區:2區TOP期刊
DOI: 10.1186/s12967-025-07027-8
我們的研究揭示了已確定的癌症易感性基因、炎症途徑和潛在候選易感性基因中的相關遺傳變異,包括BRCA2 c.1794_1798del,RAD54L基因中的1種新突變 (c.1712T>C)和 IFNAR2 基因中的c.555_559del。鑑定了ABRAXAS1、XRCC2 和 FANC 基因中的CNVs。我們還發現 RAD54L 和 MTHFR 的高表達水平與良好的生存率相關。IBC (炎性乳腺癌)的基因組成似乎非常異質。對於同一名患者,我們檢測到了幾種可能解釋疾病發展和進展的相關變異,這與在調查家庭中觀察到的癌症家族史一致。

變異(篩選)的優先級策略及流程
P: Pathogenic, LP: Likely Pathogenic, BC: Breast Cancer; MAF: Minor Allele Frequency
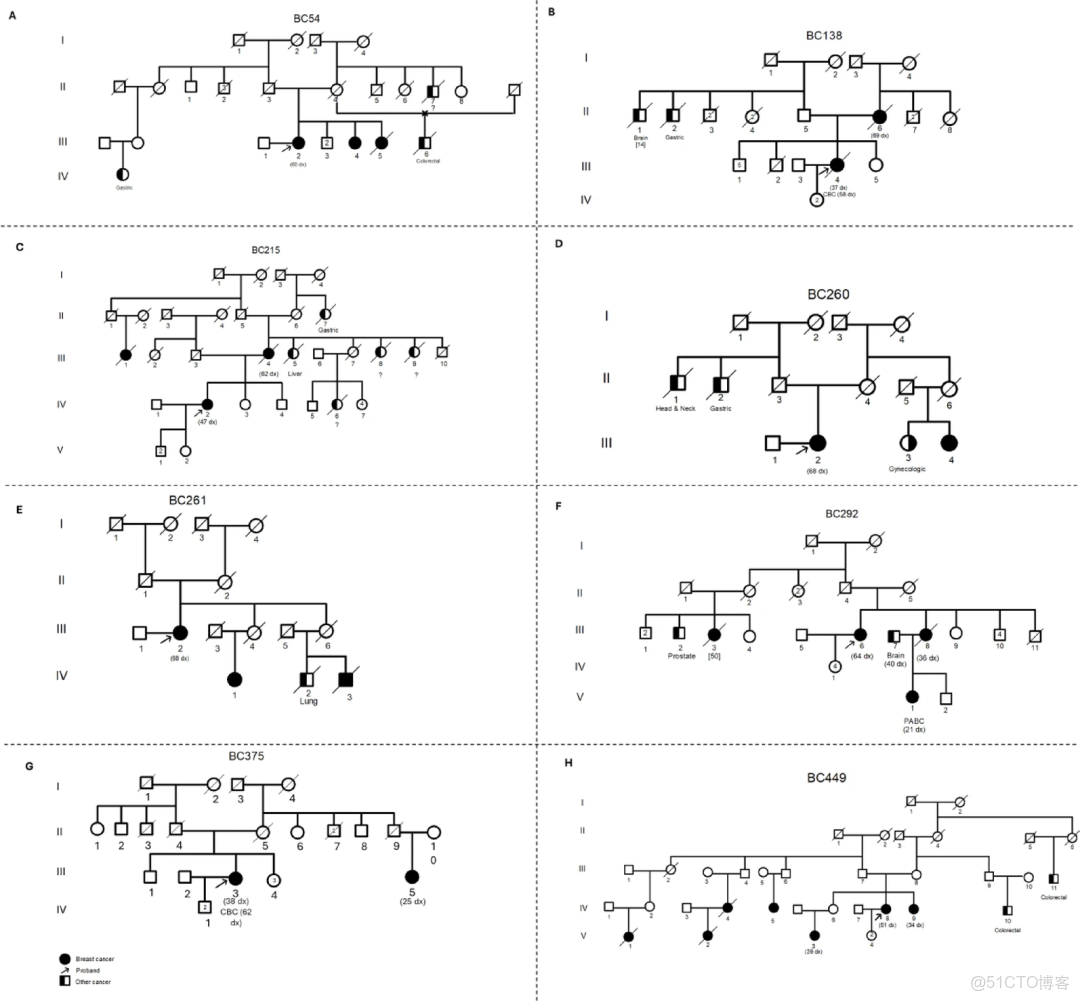
不同患者的家系圖
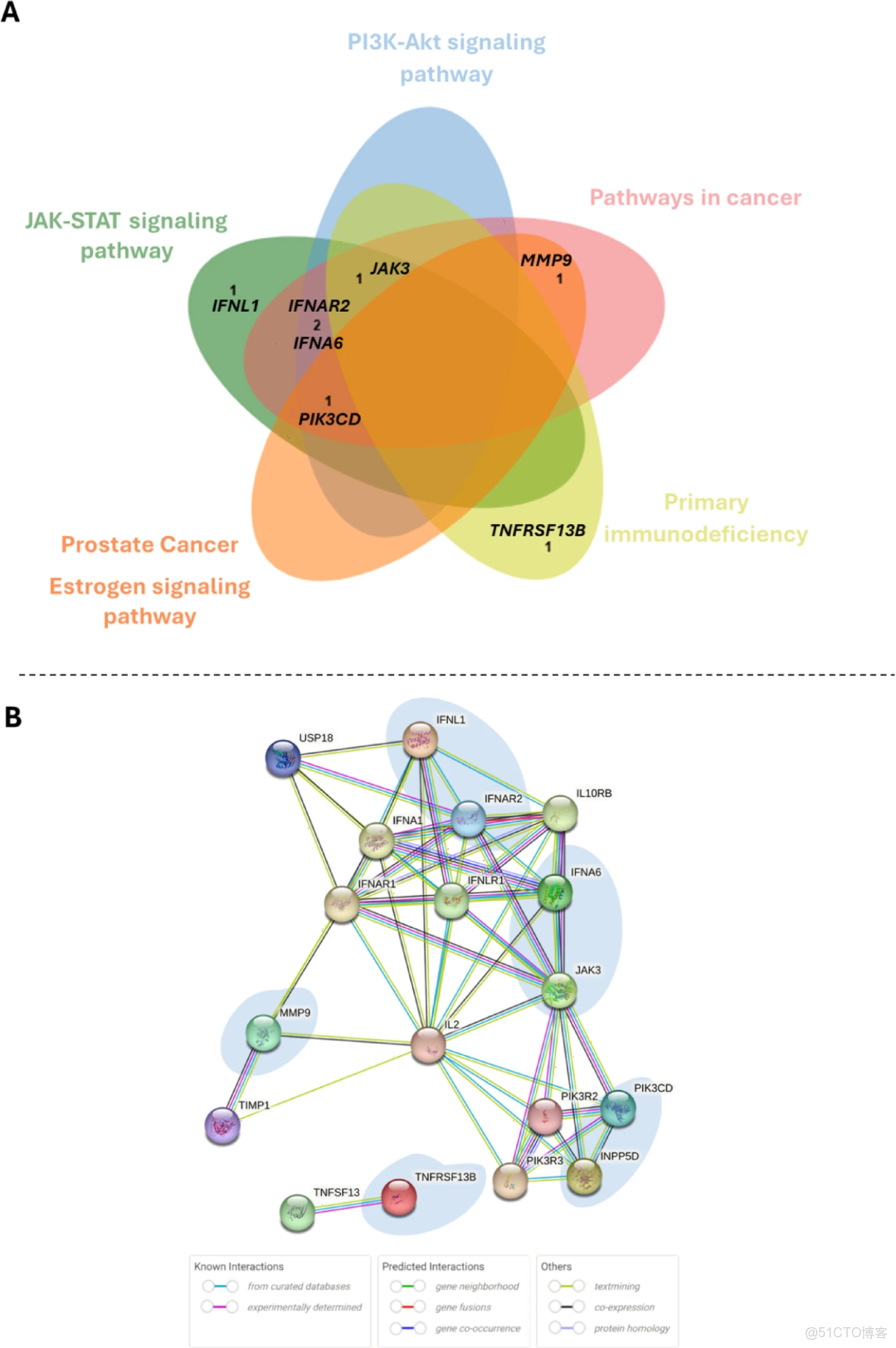
韋恩圖顯示了KEGG通路(即圖A) 和蛋白-蛋白互作網絡(即圖B),其中涉及優先的炎症及癌症相關基因。
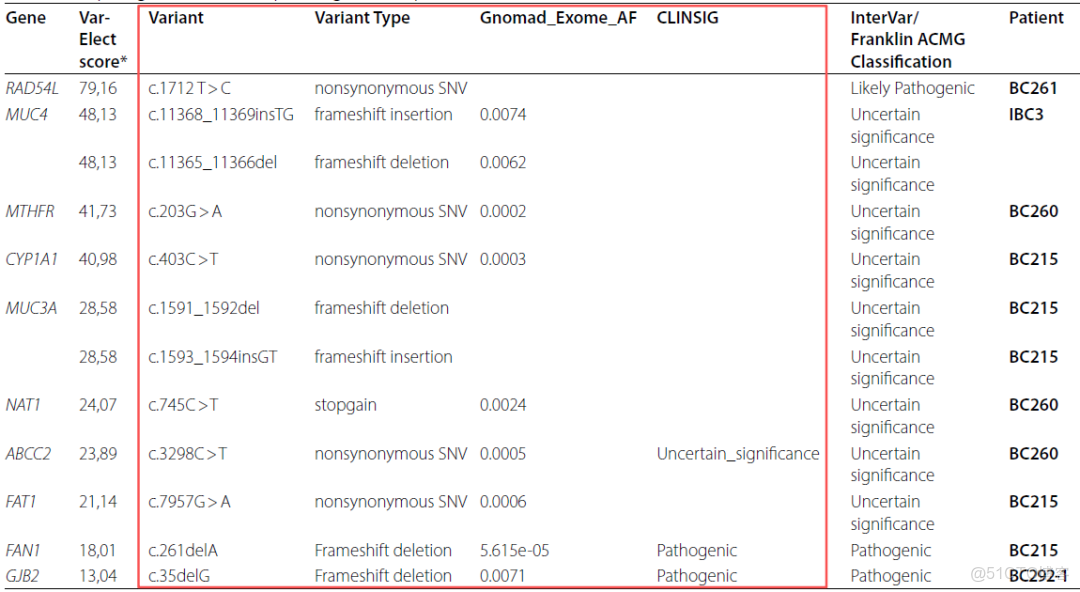
表. 根據變異致病性評分,優先排序的前10個基因及其相應變異
15. Genetic heterogeneity in pediatric short stature: insights from whole exome sequencing and snp- array analyses in a Turkish cohort
兒童身材矮小的遺傳異質性:來自土耳其隊列的全外顯子組測序和SNP芯片分析的見解
作者: Adanur Saglam K, Bekfilavioglu S, Yıldız Boyraz A, Cimbek EA, Ozden A, Turkyilmaz A, Cebi AH, Doneray H, Karaguzel G
DOI: 10.1007/s00431-025-06529-3
16. [Clinical characteristics and genetic analysis of a case with 47,XYY Disorder of sex development due to variant of NR5A1 gene]
NR5A1基因變異致1例 47,XYY 性發育異常的臨牀特徵及遺傳學分析
作者: Liu Y, Li J, Xu Q, Yang Y, He L, Duan H
DOI: 10.3760/cma.j.cn511374-20250224-00103
17. Expanding the clinical spectrum of pediatric ataxia-telangiectasia: a case series of novel genetic variants, lupus vulgaris, and hyper-IgM phenotypes
擴展兒童共濟失調-毛細血管擴張症的臨牀譜系:新遺傳變異、尋常狼瘡和高IgM表型的病例系列
作者: Bakır DB, Atay Ö, Yağmur H, Kabadayı G, Kocabey M, Asilsoy S, Uzuner N
DOI: 10.1186/s13023-025-03942-7
18. Genetic Abnormalities in Neurodevelopmental Disorders with Multidimensional Impairment
多維損傷神經發育障礙中的遺傳異常
作者: Hanin C, Torres P, Millet I, Matos J, Cravero C, Giannitelli M, Pellen AS, Pellerin H, Grossard C, Zammouri I, De Foucaud A, Laurent-Levinson C, Cohen D
DOI: 10.1007/s12031-025-02424-6
19. Evaluating gene variations in autosomal dominant polycystic kidney disease patients using whole exome sequencing and phenotype to genotype analysis
使用全外顯子組測序和表型-基因型分析評估常染色體顯性多囊腎病患者基因變異
作者: Aypek H, Balaban RF, Huriyet N, Bulut E, Cecener G, Akgur S, Gorukmez O, Unal U, Ozkaya G, Ersoy A, Oruc A, Gul CB, Yildiz A
DOI: 10.1080/0886022X.2025.2547306
20. A novel NOTCH1 nonsense variant in a bicuspid aortic valve family with intrafamilial clinical heterogeneity
一個具有家族內臨牀異質性的二葉式主動脈瓣家族中的新型NOTCH1無義變異
作者: Chen Q, Xu ZY, Chi W, Chen X, Zou J, Chen T, Wu MQ, Ruan DD, Yu HP, Zhang JH, Gao MZ, Zhang L, Liao LS, Fang ZT, Lin F, Luo JW, Chen L
DOI: 10.1186/s12872-025-05100-0
21. A Review of Syndromic Forms of Obesity: Genetic Etiology, Clinical Features, and Molecular Diagnosis
綜合徵型肥胖的綜述:遺傳病因學、臨牀特徵和分子診斷
作者: Farzand A, Rohin MAK, Awan SJ, Sharif Z, Yaseen A, Ahmad AMR
DOI: 10.3390/cimb47090718
22. A novel phex gene variant causes non-syndromic tooth agenesis
新型PHEX基因變異導致非綜合徵型牙齒缺失
作者: Pan Y, Hua B, Wang H, Tan S, Lu T, Xiong F, Ma D
DOI: 10.1186/s12903-025-06827-0
23. Genetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations
一個由SCN4A和SPG7雙突變導致骨骼肌離子通道病和遺傳性痙攣性截癱7型家系的遺傳學分析
作者: Yu HP, Xu ZY, Wu MQ, Chen Q, Zheng ZH, Wei W, Lin P, Zou J, Zhang JH, Ruan DD, Wang RL, Chen L, Gao MZ, Zhang L, Liao LS, Lin F, Li H, Fang ZT, Wang W, Ruan XL, Luo JW, Li YF
DOI: 10.1016/j.gene.2025.149782
24. Genetic determinants of the complement and coagulation pathways in invasive meningococcal disease
侵襲性腦膜炎球菌病中補體和凝血途徑的遺傳決定因素
作者: Bellos E, van Leeuwen K, Duret A, Hodeib S, Mashbat M, Kohlfuerst DS, Boeddha NP, Schlapbach LJ, Wright VJ, Fink CG, van der Flier M, van Deuren M, Sprong T, López-Trascasa M, López-Lera A, Martinón-Torres F, Salas A, Santillo D, Zenz W, Driessen GJ, Anderson ST, Secka F, Paulus S, de Groot R, Emonts M, Carrol ED, Herberg J, Levin M, Sancho-Shimizu V, Kuijpers T; EUCLIDS Consortium
DOI: 10.1016/j.jaci.2025.09.011
25. Discovery of a Novel CHD7 CHARGE Syndrome Variant (c.502C>T) by Prenatal Diagnostic Analysis: A Case Report
通過產前診斷分析發現新型CHD7 CHARGE綜合徵變異(c.502C>T):病例報告
作者: Wang K, Lin L, Fang L, Zeng L, Lin Y
DOI: 無 (僅提供PMID: 40962455)
CHARGE綜合徵:一組以記憶缺損,心臟缺損,後鼻孔閉鎖,生長髮育遲緩,生殖器畸形,耳異常為特徵的綜合徵
26. [Family Study and Blood Transfusion of a Patient with Hereditary Coagulation Factor XI Deficiency]
一例遺傳性凝血因子XI缺乏症患者的家系研究與輸血治療
作者: Han YX, Ren Y, Zhao R, Qu AC, Yang ZG
DOI: 10.19746/j.cnki.issn.1009-2137.2025.04.035
27. Genetic insights and clinical outcomes in fetal ventriculomegaly: A retrospective analysis
胎兒腦室擴張的遺傳學見解和臨牀結局:一項回顧性分析
作者: Zeng L, Yi M, Wang T, Hu J, Li H, Zhang C, Yao Y, Liu N, Qin Y, Xu R, Liu L, Song J
DOI: 10.1016/j.tjog.2025.05.016
28. Identification of a novel variant in MYRF gene in a patient with 46, XX disorders of sex development
在一名 46, XX 性發育異常患者中鑑定出MYRF基因新變異
作者: Ding L, Tian Q
DOI: 10.1080/09513590.2025.2546985
29. Identification of known and novel genetic variants in sensorineural hearing loss: insights from whole exome sequencing in Indian families
感音神經性聽力損失中已知和新遺傳變異的鑑定:來自印度家庭全外顯子組測序的見解
作者: Jagannath K, Bhat N, Ghosh A, Tawade H, Cherian S, Shettigar S
DOI: 10.1007/s11033-025-10996-0
30. Compound Heterozygous Structural Variants in Cases with Unsolved PRKN-Associated Parkinson's Disease
未解PRKN相關帕金森病病例中的複合雜合結構變異
作者: Fant A, Trova S, Monfrini E, Treves G, Musacchia F, Landuzzi F, Mandich P, Amoroso A, Sanges R, Pandolfini L, Cavallieri F, Valzania F, Fioravanti V, Di Rauso G, Brescia G, Valente EM, Tiranti V, Romito LM, Reale C, Garavaglia B, Elia AE, Cavalli A, Di Fonzo A, Vecchi M, Gustincich S
DOI: 10.1002/mds.70027
31. Functional and clinical insights into nuclear receptor variants for advancing precision diagnostics in male infertility
核受體變異的功能和臨牀見解以推進男性不育的精準診斷
作者: Gaikwad AS, Wyrwoll MJ, Koser SA, Emich J, Kuß J, Aravina M, Krallmann C, Gromoll J, Kliesch S, Laurentino S, Stallmeyer B, Friedrich C, Tüttelmann F
DOI: 10.1016/j.ebiom.2025.105899
32. Expanding the Genetic and Clinical Spectrum of Hereditary Angioedema with Normal C1 Inhibitor: Novel Variants and Treatment Insights
擴展C1抑制劑正常的遺傳性血管性水腫的遺傳和臨牀譜系:新變異和治療見解
作者: Gao H, Zhao Y, Chen S, Zhang Z, Yang F, Chen Z, Wang L, Yang J, He S, Tang C, Zheng S, Guan C, Xu Y, Tang L, Zhang A, Maurer M, Lee D, Ma L, Luo X
DOI: 10.1007/s10875-025-01912-z
33. Variants of NLRP genes encoding subcortical maternal complex components are linked to biparental placental mesenchymal dysplasia
編碼皮層下母體複合物組分的NLRP基因變異與雙親胎盤間質發育不良相關
作者: Murase A, Mishima H, Aoki S, Hara S, Kubiura-Ichimaru M, Ohba T, Yoshiura KI, Soejima H
DOI: 10.1186/s40246-025-00814-w
34. Genomic and Immune Landscape of Pancreatic Ductal Adenocarcinoma Associated with Germline Pathogenic Variants in ATM
與ATM胚系致病變變相關的胰腺導管腺癌的基因組和免疫圖景
作者: Yadav S, Bao R, Graham RP, Hu C, Hart SN, Na J, Boddicker N, Gnanaolivu RD, Smadbeck J, Ding L, Billadeau DD, Mayer AT, Majumder S, Morais Lyra PC, Lee AV, Monteiro AN, Villasboas JC, McWilliams R, Couch FJ
DOI: 10.1158/1078-0432.CCR-24-4120
35. Divergent Clonal Evolution and Early Dissemination Promote Genetic Heterogeneity of Metastases in Castration-Resistant Prostate Cancer
不同的克隆進化和早期播撒 促進去勢抵抗性前列腺癌轉移的遺傳異質性
作者: Hosseini N, Mannan R, Rebernick RJ, Su F, Wang R, Cao X, Lako A, Mellacheruvu D, Hu J, Alumkal JJ, Reichert ZR, Malik R, Mehra R, Chinnaiyan AM, Cieslik MP
DOI: 10.1158/0008-5472.CAN-24-3687
36. Case Report of Nephrogenic Diabetes Insipidus with a Novel Mutation in the AQP2 Gene
伴有AQP2基因新突變的腎性尿崩症病例報告
作者: Padilla-Guzmán A, Ochoa-Jiménez VA, Forero-Delgadillo JM, Apraez-Murillo K, Pachajoa H, Restrepo JM
DOI: 10.3390/ijms26157415
37. Exploring the unique characteristics of genes with dual autosomal dominant and recessive inheritance: mechanisms, phenotypes and candidate identification
探索具有常染色體顯性和隱性雙重遺傳模式的基因的獨特特徵:機制、表型和候選基因鑑定
作者: Ezer S, Sido T, Rips J, Hoffman Lipschuetz R, Fuchs A, Abu-Libdeh B, Chervinsky E, Damseh NS, Danial-Farran N, Morani I, Saada A, Al-Raqad M, Salah S, Yanovsky-Dagan S, Samra N, Mandel H, Shalev SA, Mor-Shaked H, Zlotogora J, Harel T
DOI: 10.1136/jmg-2025-110872
38. [Clinical and genetic analysis of a child with Primary ciliary dyskinesia variants and co-existence of CCDC39 gene variants and 22q11.21 deletion]
一例原發性纖毛運動障礙變異並CCDC39基因變異與22q11.21缺失共存的兒童臨牀及遺傳學分析
作者: Chang J, Zhang X, Han J, Wang W, Wang W, Liu L
DOI: 10.3760/cma.j.cn511374-20250521-00313
39. [Analysis of clinical manifestations and genetic variants among 11 Chinese pedigrees affected with Leber congenital amaurosis]
11箇中國Leber先天性黑蒙家系的臨牀表現和遺傳變異分析
作者: Bai Z, Shao J, Kong X
DOI: 10.3760/cma.j.cn511374-20241218-00664
Leber 先天性黑蒙症:一種嚴重的遺傳性視網膜病變,常在嬰幼兒時期發病,並伴有糖尿病、肥胖和尿崩症等一系列併發症。
40. [Newborn screening, clinical characteristics and genetic variant analysis of Glutaric acidemia type I in Henan Province]
河南省I型戊二酸血癥的新生兒篩查、臨牀特徵及遺傳變異分析
作者: Zhu X, Zhao D, Xu Y, Zhang J, Li X, Liu S, Ni M, Ren Y, Zhang C, Guo Y, Li J, Lyu S, Jia C, Shi Y
DOI: 10.3760/cma.j.cn511374-20240308-00161
41. Phenotypic spectrum of cardiac conduction disturbance and cardiomyopathy linked to titin canonical splice-site variants
與肌聯蛋白經典剪接位點變異相關的心臟傳導障礙和心肌病的表型譜
作者: Ishikawa T, Kimoto H, Seki A, Shirai M, Uto K, Makiyama T, Kitai T, Mishima H, Trujillano D, Simonet F, Baron E, Lindenbaum P, Kyndt F, Goudal A, Fukushima N, Fujita T, Hatakeyama K, Hagiwara N, Yoshiura KI, Redon R, Dina C, Estivill X, Ossowski S, Courtheix M, Probst V, Barc J, Schott JJ, Makita N
DOI: 10.1093/cvr/cvaf135
42. A Novel LAMA2 Mutation (c.7412G>A) Was Found in a Chinese Patient With Congenital Muscular Dystrophy
在一名中國先天性肌營養不良症患者中發現新型LAMA2突變(c.7412G>A)
作者: Zhao M, Liu Y, Fan L, Liu Z, Deng Y, Tao L
DOI: 10.1111/jcmm.70667
43. c.7156C > T p.(Gln2386*) variant causes loss-of-function of the USP9X gene in a female-restricted X-linked syndromic intellectual disability: a case report
c.7156C > T p.(Gln2386*) 變異 導致受限於女性的X連鎖綜合徵性智力障礙中USP9X基因功能喪失:病例報告
作者: da Silva Campos TA, Bernardes AH, Pinto IP, da Silva Teixeira HA, da Silva JF, do Prado Santos VC, Zatarin R, da Cruz AD
DOI: 10.1186/s13256-025-05456-z
44. EMC10 Gene Variants May Cause Dual Molecular Effects on the Neuropsychiatric Disease Pattern
EMC10基因變異可能對神經精神疾病模式產生雙重分子效應
作者: Bolat H, Genç Akdağ D, Ünsel-Bolat G
DOI: 10.1002/dneu.22994
45. Uniparental disomy leads to a novel cause of MC2R-related familial glucocorticoid deficiency type 1
單親二體導致MC2R相關家族性糖皮質激素缺乏症1型的新病因
作者: Müller-Nedebock AC, Wenzel E, Pfäffle R
DOI: 10.1093/ejendo/lvaf152
46. Genetic findings of children with congenital heart diseases using chromosomal microarray and trio-based whole exome sequencing
使用染色體微陣列和三人組家系全外顯子組測序對先天性心臟病兒童的遺傳學發現
作者: Guo R, Duan C, Zarrei M, Reuter MS, Dong R, Zhang G, Yang X, Zhang H, Wang Y, Scherer SW, Liu Y, Gai Z
DOI: 10.1038/s41598-025-06977-9
47. HECW2 Gene Mutation: A Rare Cause of West Syndrome: A Case Report
HECW2基因突變:West綜合徵的罕見病因:病例報告
作者: Meena AK, Mahesan A, Kamila G, Jauhari P, Chakrabarty B, Kumar A, Gulati S
DOI: 10.4103/neurol-india.Neurol-India-D-23-00203
West綜合徵,又稱嬰兒痙攣,是嬰兒期常見的癲癇性腦病,以頻繁強直痙攣發作、腦電圖高峯失律及智力運動發育障礙為特徵。
48. [Clinical features and molecular pathogenesis of neurodevelopmental disorder with impaired speech and hyperkinetic movements associated with ZNF142 gene variants]
ZNF142基因變異相關伴語言障礙和運動亢進的神經發育障礙的臨牀特徵及分子發病機制
作者: Xu Y, Zhao XK, Xuan XY
DOI: 10.3760/cma.j.cn112140-20250519-00430
49. AUTS2 disruption underlies radioulnar synostosis and skeletal dysmorphogenesis: evidence from four unrelated cases
AUTS2破壞是橈尺骨融合和骨骼形態發生異常的基礎:來自4個獨立病例的證據
作者: Liu C, Shen F, Deng M, Yang C, Zhao L, Zhu G, Wang H, Li Z, Yang Y
DOI: 10.1136/jmg-2025-110886
50. Genetic etiology of 283 Chinese individuals with epilepsy using copy number variation sequencing and whole exome sequencing: a single-center cohort study
使用拷貝數變異測序和全外顯子組測序對283名中國癲癇患者的遺傳病因學研究:一項單中心隊列研究
作者: Hu J, Xin M, Liu J, Li H, Li X, Chen L, Yang P, Zhao H, Sun P, Gao G, Feng H, Li Z, Xiao G, Li Y, Li K, Xu X
DOI: 10.1186/s12920-025-02184-7
51. Aryl hydrocarbon receptor interacting protein and syndromic gene variants detected in Turkish isolated pituitary adenoma families by whole exome sequencing
通過全外顯子組測序在土耳其孤立性垂體腺瘤家系中檢測到芳香烴受體互作蛋白及綜合徵性基因變異
作者: Ertorer ME, Tuncer FN, Ciftci S, Tanrikulu S, Selcukbiricik OS, Topaloğlu Ö, Evran M, Kadioglu P, Aydin S, Can B, Sehit C, Pekkolay Z, Oruk GG, Cetinarslan B, Yarman S
DOI: 10.1038/s41598-025-08610-1
52. [Clinical and genetic analysis of a patient with unilateral Pigmented paravenous retinochoroidal atrophy and Retinitis pigmentosa in the contralateral eye related to CRB1 gene variant]
CRB1基因變異相關單眼靜脈旁視網膜脈絡膜萎縮伴對側眼視網膜色素變性1例患者的臨牀表型及遺傳學分析
作者: Tang Y, Huang H, Lin X, Chi Z
DOI: 10.3760/cma.j.cn511374-20241108-00580
53. [Clinical phenotype and genetic analysis of four cases of Epileptic encephalopathy caused by PCDH19 mutations]
四例PCDH19突變所致癲癇性腦病的臨牀表型及遺傳學分析
作者: Wei L, Wang J, Yao R, Wang J, Yu T
DOI: 10.3760/cma.j.cn511374-20241009-00523
54. Primary pigmented papillary epithelial tumor of the sella: case report and literature review
鞍區原發性色素性乳頭狀上皮腫瘤:病例報告及文獻綜述
作者: Wu S, Yang X, Wang X
DOI: 10.1007/s10014-025-00508-0
55. Whole exome-based variant profiling and functional network characterization in neural tube defects
神經管缺陷中基於全外顯子組的變異分析和功能網絡表徵
作者: Akcali N, Yildiz SH, Erdogan MO, Eslamkhah S, Elmas M, Pehlivan A, Solak M
DOI: 10.1007/s00381-025-06884-4
56. Genetic etiology of ventriculomegaly in 73 fetuses identified by High-Throughput sequencing
通過高通量測序鑑定73例胎兒腦室擴張的遺傳病因
作者: Chenyue Z, Huiqin X, Jingbo G, Min G, Hao Y, Rong G, Guizhi C, Xiayu S, Jianrui W
DOI: 10.1038/s41598-025-06714-2
57. Etiological diagnosis of miscarriage by combining use of chromosomal microarray analysis and whole-exome sequencing
聯合使用染色體微陣列分析和全外顯子組測序進行流產的病因學診斷
作者: Zhuang J, Fu W, Gu L, Ye X, Wang J, Chen C
DOI: 10.1186/s40001-025-02709-x
58. Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome
擴展TCTN3相關Joubert綜合徵的基因型和表型譜
作者: Lo Giudice M, Borgione E, Giuliano M, Santa Paola S, Di Blasi FD, Pettinato R, Romano C, Scuderi C
DOI: 10.3390/genes16060706
Joubert綜合徵是由Joubert等於1969年首先報道,是一種較罕見的發育畸形,多數為常染色體隱性遺傳病。典型神經病理學改變為小腦蚓部發育不良或不發育,齒狀核、腦橋基底部及延髓的神經核團也可發育不良,錐體交叉幾乎完全缺如。患兒年齡很小即可出現症狀,表現為肌張力減低,共濟失調,運動及智力發育落後,間歇性呼吸深快及眼球運動異常。文獻報道患病率約為1/10萬,男女之比約3:2。
59. Caveolin 3 Variant T78M in a Large Family With Brugada Syndrome: Clinical Features and Coexistence of ADRB1 and GRK5 Gene Mutation
一個Brugada綜合徵大家系中的Caveolin 3變異T78M:臨牀特徵及ADRB1和GRK5基因突變的共存
作者: Santoro F, D'Apolito M, Ragnatela I, Ranaldi A, Margaglione A, D'Alessandro D, Niglio FP, Santacroce R, Cannito S, D'Andrea G, Pellegrino P, Mazzanti A, Di Biase L, Margaglione M, Brunetti ND
DOI: 10.1111/jce.16770
Brugada綜合徵是由於編碼心肌離子通道基因的突變,引起離子通道功能異常而導致的綜合徵。臨牀上,這個綜合徵以V1~3導聯ST段抬高、V1~3導聯ST段多變、心臟結構無明顯異常、多形室性心動過速(室速)或心室顫動(室顫)和暈厥的反覆發作、以及心臟性猝死為特徵。
60. The hidden causes of pregnancy loss: a closer look
妊娠丟失的隱藏原因:深入觀察
作者: Shi P, Wang C, Liang H, Zhu X, Wang X, Ning Y, Leigh D, Cram DS, Kong X
DOI: 10.1186/s12967-025-06678-x
61. Structural variation in nebulin and its impact on phenotype and inheritance: establishing a dominant distal phenotype caused by large deletions
伴肌動蛋白的結構變異及其對錶型和遺傳的影響:確立由大片段缺失引起的顯性遠端表型
作者: Sagath L, Kiiski K, Naidu K, Patel K, Jonson PH, Laarne M, Djordjevic D, Yoon G, LaGroon A, Rogers C, Galindo MK, Scherer K, Kunstmann E, Koparir E, Ho D, Davis M, Joshi P, Zygmunt A, Orbach R, Donkervoort S, Bönnemann CG, Savarese M, Echaniz-Laguna A, Biancalana V, Genetti CA, Iannaccone ST, Beggs AH, Wallgren-Pettersson C, Henning F, Pelin K, Lehtokari VL
DOI: 10.1038/s41431-025-01891-0
62. Whole exome sequencing unravels genetic architecture and its clinical implications in pediatric pulmonary arterial hypertension
全外顯子組測序揭示兒童肺動脈高壓的遺傳結構及其臨牀意義
作者: Jiang DJ, Yang YJ, Wang YZ, Zhang X, Chan WX, Yu TT, Chen H, Zhang H, Yan Y, Fu LJ
DOI: 10.1016/j.ijcard.2025.133515
63. Long-Read Sequencing Identifies Mosaic Sequence Variations in Friedreich's Ataxia-GAA Repeats
長讀長測序鑑定弗裏德賴希共濟失調GAA重複序列中的嵌合序列變異
作者: Park J, Dufke C, Fleszar Z, Schlotterbek M, Buena-Atienza E, Stühn LG, Gross C, Sturm M, Ossowski S, Schöls L, Riess O, Haack TB
DOI: 10.3390/ijms26114969
64. [Diagnosis, treatment, and genetic analysis of five cases of primary atypical hemolytic uremic syndrome]
五例原發性非典型溶血尿毒綜合徵的診斷、治療及遺傳學分析
作者: He WY, Tian F, Li J, Han RH, Xing GQ
DOI: 10.3760/cma.j.cn112138-20241209-00807
65. Application of RNA-seq for single nucleotide variation identification in a cohort of patients with hypertrophic cardiomyopathy
應用RNA測序在一組肥厚型心肌病患者中進行單核苷酸變異鑑定
作者: Chumakova A, Vlasov I, Filatova E, Klass A, Lysenko A, Salagaev G, Shadrina M, Slominsky P
DOI: 10.1038/s41598-025-03226-x
66. Predicting expression-altering promoter mutations with deep learning
使用深度學習預測改變表達的啓動子突變
作者: Jaganathan K, Ersaro N, Novakovsky G, Wang Y, James T, Schwartzentruber J, Fizhev P, Kassam I, Cao F, Hawe J, Cavanagh H, Lim A, Png G, McRae J, Banerjee A, Kumar A, Ulirsch J, Zhang Y, Aguet F, Wainschtein P, Sundaram L, Salcedo A, Panagiotopoulou SK, Aghamirzaie D, Padhi E, Weng Z, Dong S, Smedley D, Caulfield M, O'Donnell-Luria A, Rehm HL, Sanders SJ, Kundaje A, Montgomery SB, Ross MT, Farh KK
DOI: 10.1126/science.ads7373
67. Different mutations in TBL1XR1 lead to diverse phenotypes of neurodevelopmental disorder: two case reports
TBL1XR1的不同突變導致神經發育障礙的多種表型:兩例病例報告
作者: Wei L, Yang Y, Jiang T, Zhang C, Chen C, Huang M, Li N, Xiong H, Gao F
DOI: 10.1186/s12920-025-02169-6
68. Effects of the Missense Variants on Complete Phenotype and Splicing Variant on Severe Growth Retardation in the BPTF Gene
BPTF基因錯義變異對完全表型的影響及剪接變異對嚴重生長遲緩的影響
作者: Ünsel-Bolat G, Gerik-Celebi HB, Durgut BD, Türkyılmaz A, Bolat H
DOI: 10.1002/dneu.22970
69. Identifying the germline variation spectrum and predisposition genes in Chinese ovarian cancer using whole exome sequencing
使用全外顯子組測序鑑定中國卵巢癌患者的胚系變異譜和易感基因
作者: Guan X, Liao S, Zhang F, Zhu Q, Qiu H, Qin L, Zhang X
DOI: 10.1186/s12885-025-14302-w
70. A novel likely pathogenic variant in the mitochondrial ribosomal protein L44 (MRPL44) associated with hypertrophic cardiomyopathy in Tunisian patients
與突尼斯患者肥厚型心肌病相關的線粒體核糖體蛋白L44(MRPL44)新型可能致病性變異
作者: Gargouri R, Ammous-Boukhris N, Hssairi M, Mosbah A, Jabeur M, Feki W, Mnif Z, Mokdad-Gargouri R, Abid L, Gargouri L
DOI: 10.1007/s11033-025-10556-6
71. Genetic detection of a novel LRAT pathogenic variant in patients with early-onset severe retinal dystrophy
對早發性嚴重視網膜營養不良患者中新型LRAT致病性變異的遺傳檢測
作者: Deng WL, Liu KY, Liu SL
DOI: 10.1080/13816810.2025.2507083
72. A novel 5bp deletion in HPS4 gene associates with Hermansky-Pudlak Syndrome
HPS4基因新型5bp缺失與 Hermansky-Pudlak 綜合徵相關
作者: Premkumar S, Shiva Sankari S, Sandhra G, R Pillai M, Krishnadas SR, Sundaresan P
DOI: 10.1080/13816810.2025.2502361
Hermansky-Pudlak綜合徵(HPS) 是一種罕見的常染色體隱性遺傳病,以 血小板功能障礙、眼皮膚白化病 及 多器官累及 為特徵。 病因涉及 HPS相關基因突變,導致細胞器(如溶酶體、黑素體等)功能異常。 典型表現包括 出血傾向、視力問題、皮膚毛髮顏色淺淡,部分患者會進展為 肺纖維化、肉芽腫性結腸炎 等嚴重併發症。 診斷需結合臨牀、實驗室及基因檢測,目前 無根治方法,以對症支持治療為主。
73. Computational exploration of TITIN variations: insights from whole exome sequencing and molecular dynamics simulation study
TITIN變異的計算探索:來自全外顯子組測序和分子動力學模擬研究的見解
作者: Mukhopadhyay A, Devi B, Baidya ATK, Yadav ML, Kumar R, Mohapatra B
DOI: 10.1080/07391102.2025.2500683
74. Prenatal diagnosis and molecular cytogenetic analyses of a rare 17q12 microdeletion and 17q11.2 microduplication family with normal phenotype
一個表型正常的罕見17q12微缺失和17q11.2微重複家系的產前診斷和分子細胞遺傳學分析
作者: Tian W, Xia Q, He X, Leng P
DOI: 10.1016/j.ejogrb.2025.114023
75. Clinical differences between female monozygotic twins with X-linked Alport syndrome with somatic mosaicism
患有X連鎖Alport綜合徵伴體細胞嵌合的女性同卵雙胞胎的臨牀差異
作者: Mikami N, Kitakado H, Kimura N, Sakakibara N, Nozu K, Hamada R
DOI: 10.1007/s00467-025-06772-8
Alport綜合症:又稱眼-耳-腎綜合徵,為遺傳性腎炎中最常見一種。臨牀表現似慢性腎小球腎炎。
76. Clinical and Radiological Characterization of TEFM-Associated Neurological Disorder
TEFM相關神經系統疾病的臨牀和影像學特徵
作者: Adarsha N, Sait H, Ravichandran D
DOI: 10.1002/ajmg.a.64101
77. The improvement in diagnostic yield of developmental and epileptic encephalopathy by the multi-omics sequential testing method
多組學序貫檢測方法提高發育性和癲癇性腦病的診斷率
作者: Yang SH, Liu J, Quan Y, Lin G, Zhou X, He H, Gan X, Yang T, Cui MY, Du X, Quan X, Gu W, Zhang HY, Wang H, Guan W
DOI: 10.1016/j.bbadis.2025.167854
78. Three Novel Mutations in TUBB8 Cause Female Infertility Due to Multiple Morphological Abnormalities of the Oocyte and Early Embryo
TUBB8基因的3個新突變導致因卵母細胞和早期胚胎多形態異常引起的女性不孕
作者: Li D, Yuan G, Wang X, Zhuang J, Wang L, Liu Y, Liu X, Han L, Dou H, Li B, Hao C
DOI: 10.1007/s43032-025-01844-4
79. Adolescent-onset hyperhomocysteinaemia: cases report and literature review
青少年發病的高同型半胱氨酸血癥:病例報告及文獻綜述
作者: Li Y, Wang L, Yu L, Miao X, Zhang L, Sun S, Wang C, Sun Y
DOI: 10.1080/13554794.2025.2489928
80. Identification of Genetic Variants Causing Paediatric Cataract in Myanmar
鑑定導致緬甸兒童白內障的遺傳變異
作者: Jones JL, Boardman D, Nweni K, Edo F, Barros de Lima IM, Lertsinpakdee P, Hlaing S, Casson R, Mallipatna A, Win Y, McComish B, Lin N, Muecke J, Griffiths A, Holmes M, Aung TH, Burdon KP
DOI: 10.1111/cge.14755
81. Whole-genome and whole-exome sequencing of Mayer-Rokitansky-Küster-Hauser syndrome-discordant monozygotic twins
對不一致罹患Mayer-Rokitansky-Küster-Hauser綜合徵的同卵雙胞胎進行全基因組和全外顯子組測序
作者: Ma W, Fu F, Wang W, Ma X, Wang S, Wang M, Li Y
DOI: 10.1007/s10815-025-03440-6
MRKH綜合徵 (Mayer-Rokitansky-Küster-Hauser syndrome)是一種先天性疾病,發病率約1/4500~1/5000,主要影響女性。在胚胎髮育時,苗勒管出問題,致使患者的子宮、道等內生殖器官發育異常。 患者多表現為青春期無月經,可能有道短、閉鎖等狀況。部分人還會出現泌尿系統或骨骼異常,如腎臟畸形、脊柱側彎等。
82. The molecular landscape of hereditary ataxia: a single-center study
遺傳性共濟失調的分子圖譜:一項單中心研究
作者: Bregant E, Betto E, Dal Secco C, Zucco J, Baldan F, Allegri L, Lonigro IR, Faletra F, Verriello L, Damante G, Mio C
DOI: 10.1007/s00439-025-02744-y
83. LSM1 c.231+4A>C hotspot variant is associated with a novel neurodevelopmental syndrome: first patient cohort
LSM1 c.231+4A>C熱點變異與一種新型神經發育綜合徵相關:首個患者隊列
作者: Reytan Miron S, Kurolap A, Abu-Libdeh B, Abu-Libdeh AS, Velmans C, Erger F, Riehmer V, Hsieh TC, Lesmann H, Reches A, Chai Gadot C, Mory A, Al-Ashhab M, Netzer C, Damseh N, Baris Feldman H
DOI: 10.1136/jmg-2024-110574
84. Intellectual disability and retinitis pigmentosa due to a homozygous null SCAPER variant: a clinical and genetic insight with review of the literature
由於純合SCAPER無效變異導致的智力障礙和視網膜色素變性:臨牀和遺傳學見解及文獻綜述
作者: Manav Yiğit Z, Dikbaş OS, Erkan E, Vural GŞ, Bozkurt G
DOI: 10.1080/13816810.2025.2485222
85. Expanding the SIAH1-Associated Phenotypic Spectrum: Insights From Loss-Of-Function Variants
擴展SIAH1相關表型譜:來自功能喪失性變異的見解
作者: Douiev L, Alvarez PF, Frank M, Hanington L, Hoffman TL, Irons MB, Kim J, Kumar A, Lasa-Aranzasti A, Le Duc D, Livesey H, Murch O, Shears D, Walther BK, Harel T
DOI: 10.1002/ajmg.a.64048
86. Prenatal diagnosis and genetic counseling of a Chinese family with inherited multiple chromosomal microduplications
一箇中國遺傳性多染色體微重複家系的產前診斷和遺傳諮詢
作者: Hu F, Zhang G
DOI: 10.1097/YPG.0000000000000391
87. A Novel Heterozygous c.1024A>G Variant in BMPR1B Causes Either Isolated Brachydactyly Type A4 With Variable Expressivity or Incomplete Type A4 Overlapping Type D in a Chinese Han Pedigree
BMPR1B基因新型雜合 c.1024A>G 變異導致中國漢族家系中出現具有可變表現度的孤立性A4型短指症或不完全性A4型與D型重疊
作者: Yang X, Wu X, Li H, Zhou R, Guo K, Shang C, Zhao S, Ma M
DOI: 10.1002/ajmg.a.64060
88. De novo TANC2 variants caused developmental and epileptic encephalopathy and epilepsy
新發TANC2變異導致發育性和癲癇性腦病及癲癇
作者: Luo S, Zhang WJ, Jiang M, Ren RN, Liu L, Li YL, Liu WH, Wang PY, Gu YJ, Chen LZ, Shen LP, Tian Y, Liu XR, Yi YH, Liao WP, Zhou P; China Epilepsy Gene 1.0 Project
DOI: 10.1111/epi.18358
89. Identification and structural analysis of pathogenic variants in MYOC and CYP1B1 genes in Indian JOAG patients
印度JOAG患者MYOC和CYP1B1基因致病性變異的鑑定和結構分析
作者: Yadav M, Kumar M, Dhull CS, Sachdeva S, Bhardwaj A, Yadav A, Panghal V, Sharma P, Kumari A, Yadav R, Singh M, Kumar R, Deora A, Rathi M, Kaur P, Tanwar M
DOI: 10.1007/s10384-025-01173-8
90. TP53 Mutations and PD-L1 Amplification in Vulvar Adenocarcinoma of the Intestinal Type: Insights From Whole Exome Sequencing of 2 Cases
腸型外陰腺癌中的 TP53 突變和PD-L1擴增:來自2例全外顯子組測序的見解
作者: Fujii E, Kato MK, Ono H, Yamaguchi M, Higuchi D, Koyama T, Komatsu M, Hamamoto R, Ishikawa M, Kato T, Kohno T, Shiraishi K, Yoshida H
DOI: 10.1097/PGP.0000000000001093
91. Revealing Molecular Diagnosis With Whole Exome Sequencing in Patients With Inherited Retinal Disorders
利用全外顯子組測序揭示遺傳性視網膜疾病患者的分子診斷
作者: Yavas C, Arvas YE, Dogan M, Gezdirici A, Aslan ES, Karapapak M, Barıs S, Eroz R
DOI: 10.1111/cge.14708
92. NRXN2 Homozygous Variant Identified in a Family with Global Developmental Delay, Severe Intellectual Disability, EEG Abnormalities and Speech Delay: A new Syndrome?
在一個全面發育遲緩、嚴重智力殘疾、腦電圖異常和言語遲緩的家系中鑑定出NRXN2純合變異:一個新綜合徵?
作者: Karaer D, Özçelik AA, Karaer K
DOI: 10.1177/15500594241309948
93. Challenging the narrative of Alport syndrome spectrum: no link with cystic phenotype
挑戰Alport綜合徵譜系的敍事:與囊性表型無關
作者: Pagniez MS, Lombardi Y, Fages V, Larrue R, Laboux T, Gatinois C, Letavernier E, Rigothier C, Glowacki F, Mesnard L, Robert T
DOI: 10.1093/ndt/gfae290
Alport綜合症:遺傳性腎炎中最常見一種,臨牀表現似慢性腎小球腎炎。
94. Homozygous missense variations of APC12 cause meiotic metaphase I arrest in oocytes and female infertility
APC12的純合錯義變異導致卵母細胞減數分裂中期I停滯和女性不孕
作者: Lin Y, Wei Z, Zhang L, Yao Y, Huang Y, Yao G, Wang W, Hu S, Ding Y, Lu Y, Bian X, Dong X, Guan H, Huang Y, Sun Y
DOI: 10.1016/j.ajog.2024.11.013
95. A Novel m.1636A > G Variant in Mitochondrial TV Gene Might Cause New Phenotype of Mitochondrial Disease in a 2-Year Old Chinese Boy
線粒體TV基因新型m.1636A > G變異可能導致一名2歲中國男孩出現線粒體疾病新表型
作者: Yang H, Zhang VW, Ai L, Wu L
DOI: 10.1007/s12035-024-04472-2
96. Isolated Congenital Anosmia: Case Report and Literature Review
孤立性先天性嗅覺喪失:病例報告及文獻綜述
作者: Alotaibi NH, Alrashed M, Drad MK, Abu-Safieh L, Almobarak AA, Baz B, Farzan RA, Alsuhaibani MS, Al-Alsheikh Y
DOI: 10.1177/01455613221111496