
基於整合蛋白質組學和臨牀數據的可解釋機器學習模型預測乳腺癌5年生存率

iMetaMed主頁:https://onlinelibrary.wiley.com/journal/3066988x
研究論文
● 原文:iMetaMed
● 英文題目:An Interpretable Machine Learning Model for Predicting 5-Year Survival in Breast Cancer Based on Integration of Proteomics and Clinical Data
● 中文題目:基於整合蛋白質組學和臨牀數據的可解釋機器學習模型預測乳腺癌5年生存率
● 原文鏈接:https://onlinelibrary.wiley.com/doi/10.1002/imm3.70010
● DOI: https://doi.org/10.1002/imm3.70010
● 2025年10月7日,温州醫科大學附屬第一醫院夏二傑和王甌晨在iMetaMed在線發表了題為“An Interpretable Machine Learning Model for Predicting 5-Year Survival in Breast Cancer Based on Integration of Proteomics and Clinical Data”的研究文章。
● 本研究首次將KAN引入深度學習模型,用於整合臨牀與蛋白質組學數據以預測乳腺癌患者的5年生存率。包含13個關鍵預測因子的最優預測模型展現出卓越的預測性能,具有高精度、高精確度及F1分數。通過SHAP解釋方法,既可基於個性化輸入數據進行個體預測,又能全面解析特徵貢獻。KAN通過構建特徵輸入與預測結果之間的數學框架,提升了模型的可解釋性。該研究為最終模型的臨牀轉化應用提供了有力支持。
● 第一作者:吳志炫、姚聖楠、金玲莉、吳雪
● 通訊作者:夏二傑、王甌晨
● 合作作者:章榕榕
● 主要單位:温州醫科大學附屬第一醫院乳腺外科、浙江大學醫學院附屬邵逸夫醫院結直腸外科、同濟大學醫學院附屬同濟醫院乳腺外科
亮點
● 我們的模型在預測乳腺癌5年生存率方面展現出穩健性能;
● SHAP分析識別出影響模型預測的關鍵特徵;
● KAN通過提供數學函數表達式對模型進行了優化。
摘 要
乳腺癌是全球女性中高度異質性的惡性腫瘤。僅依靠臨牀病理特徵的傳統預後模型預測準確性有限且缺乏分子水平的見解。與這些傳統方法不同,本研究將蛋白質組學與臨牀數據整合至可解釋深度學習框架中,以提升預後精確度和生物學可解釋性。我們旨在利用多組學數據建立一個更可靠的模型來準確預測乳腺癌患者的5年生存狀態。與其他特徵組合模型相比,整合蛋白質組學與臨牀特徵的模型(AUC=0.8136)表現出更優越的性能。經優化的13個關鍵特徵模型(4個臨牀特徵與9個蛋白)實現了0.864的AUC值,精確度0.970,召回率0.810,F1分數0.883。SHapley加性解釋(SHAP)分析確定MPHOSPH10、EGFR、ARL3、KRT18、淋巴結狀態和HER2狀態為最具影響力的特徵,而科爾莫戈洛夫-阿諾爾德網絡(KAN)分析則揭示了關鍵貢獻因素與預測結果間的明確數學關係。總的來説,我們開發的可解釋多模態模型在預測乳腺癌患者5年生存率方面表現出強大性能,並提供了機制上的見解,通過開發易用預測工具增強了其臨牀轉化潛力。
視頻解讀
Bilibili:https://www.bilibili.com/video/BV1CyUrB5EBW/
Youtube:https://youtu.be/2dup5feL1Zc
中文翻譯、PPT、中/英文視頻解讀等擴展資料下載
請訪問期刊官網:http://www.imeta.science/
全文解讀
引 言
乳腺癌仍是全球女性中最常見的惡性腫瘤之一,具有複雜的生物學特徵和多樣化的臨牀結局。儘管診斷和治療策略取得了進展,但由於腫瘤異質性和個體治療反應的差異性,患者生存期的準確預測仍具挑戰性。傳統的預後模型主要依賴腫瘤大小、淋巴結狀態和激素受體表達等臨牀病理特徵,但這些參數單獨使用對患者結局的預測準確性有限。因此,開發一個更穩健的多組學預後模型來預測乳腺癌患者的5年生存率具有重要意義。
多組學技術的最新進展使得對腫瘤進行全面分子分析成為可能,包括基因組學、轉錄組學和蛋白質組學。這些高通量方法為腫瘤生物學和潛在預後生物標誌物研究提供了前所未有的洞見。特別是,蛋白質組學能夠直接反映細胞功能與表型,可能比單獨的基因組或轉錄組標記物更能可靠地指示疾病進展和治療反應。先前的研究表明,蛋白質組學分析可為乳腺癌提供有價值的預後信息。例如,反相蛋白質陣列分析已鑑定出與無復發生存期相關的蛋白質特徵,大規模蛋白質組學研究也定義了具有不同生存結局的分子亞型。然而,這些研究通常單獨評估蛋白質組學特徵或在轉錄組數據背景下進行分析,而未將其與臨牀預後因素整合到一個統一的預測模型中。據我們所知,目前尚未有公開發表的研究開發出將蛋白質組學特徵與常規收集的臨牀特徵相結合的預後模型,用以預測乳腺癌患者的5年生存率。
人工智能(AI),尤其是深度學習算法,在整合與分析複雜的多維生物醫學數據方面展現出顯著潛力。這些計算方法能夠識別大規模數據集中傳統統計方法難以察覺的複雜模式與關聯。然而,AI模型的臨牀實用性常受限於其"黑箱"特性,導致臨牀醫生難以理解和信任預測依據。可解釋人工智能(XAI)作為AI研究的新興前沿領域,專門應對機器學習(ML)算法"黑箱"的可解釋性挑戰。在各種XAI方法中,SHapley加性解釋(SHAP)已成為模型解讀的強效技術。與局部可解釋模型無關解釋(LIME)等廣泛應用的局部解釋方法相比,科爾莫戈洛夫-阿諾爾德網絡(KAN)在生物醫學應用中具有多項優勢。KAN可直接學習輸入特徵與目標結果間的顯式函數關係,實現線性和非線性效應的可視化與量化。這種全局可解釋性避免了LIME依賴單個預測擾動可能產生的不穩定性與局部偏差。
本研究旨在開發並驗證一種透明、可解釋的人工智能模型,該模型整合蛋白質組學與臨牀數據,用於預測乳腺癌患者的5年生存率。通過採用SHAP和KAN等先進可解釋性技術,我們試圖闡明驅動預測結果的生物學和臨牀因素。此外,我們旨在開發一個基於網絡的直觀工具,通過實時預測與特徵貢獻度的全面可視化,提升臨牀轉化應用價值。最後,通過免疫組織化學染色驗證關鍵蛋白的表達情況。
結 果
患者特徵
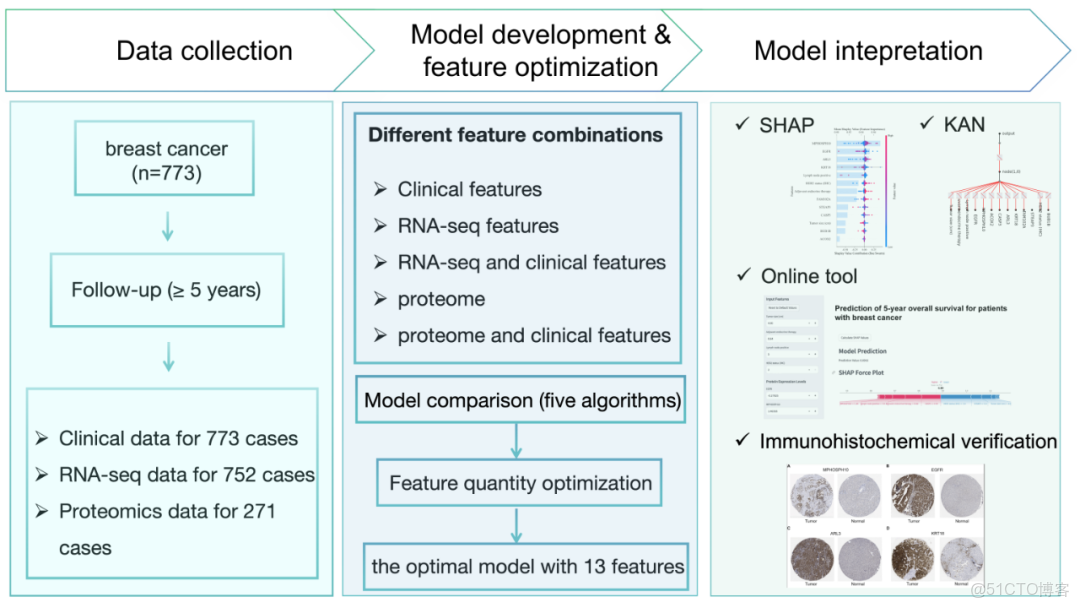
本研究共納入773名乳腺癌患者。訓練集與測試集的基線特徵(補充資料:表8)包括年齡、絕經狀態、組織學類型、組織學分級、淋巴結陽性等人口統計學特徵。中位隨訪時間為83.1個月。研究設計如圖1所示。
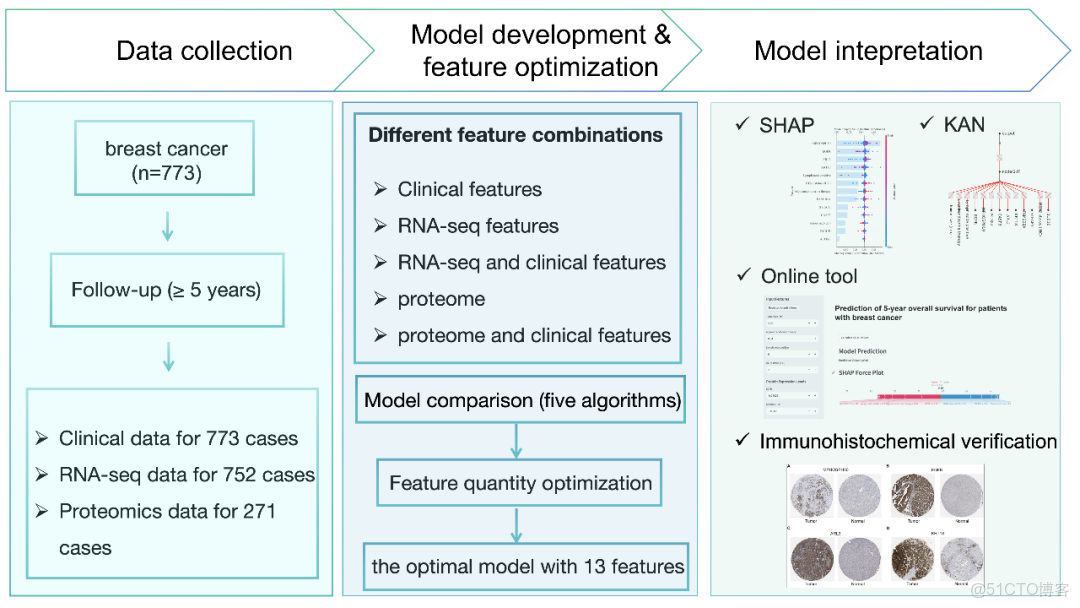
圖1. 研究的工作流程
模型開發與特徵優化
為確定預測乳腺癌患者5年生存率(是或否)的最佳多組學特徵,我們採用深度神經網絡(DNN)構建了五種特徵組合方案。在測試集中AUC值為0.624(臨牀特徵),0.716(RNA測序特徵),0.711(RNA測序聯合臨牀特徵),0.720(蛋白質組)和0.814(蛋白質組聯合臨牀特徵),如圖2A所示。其中蛋白質組與臨牀特徵的組合模型展現出最優預測性能。此外,模型評價指標進一步證實了蛋白質組與臨牀特徵組合模型的優越性能:準確率0.811、召回率0.861、精確率0.919和F1分數0.889(補充資料:圖1A)。
為提高計算效率並降低維度,我們採用三步特徵篩選策略(圖2B)。首先,基於過濾器的方法將特徵池縮減至100個候選指標;其次,採用嵌入式方法將其縮小到50;最後,基於包裝器的技術確定了20個最具信息的變量。該過程中模型性能保持穩定,其中20個特徵的模型達到最高準確率(0.890)和F1分數(0.849),表明在不損失預測能力的前提下可大幅降維。隨後使用五種機器學習算法評估這20個特徵(圖2C),其中DNN表現最佳(AUC=0.877),優於XGBoost(0.644)、邏輯迴歸(0.792)、KNN(0.643)和樸素貝葉斯(0.585)(補充資料:圖1B)。隨後,採用SHAP值優化特徵(圖2D)。性能隨着更高等級特徵的加入而提升,但在13個特徵後趨於穩定。13個特徵的模型AUC達0.864,與20個特徵模型(0.877)相當,同時更具簡約性。精確率(0.970)、召回率(0.810)和F1分數(0.883)進一步證實其穩健預測能力(補充資料:圖1C)。因此,最終選取前13個特徵用於後續分析,包括4個臨牀變量(腫瘤大小、輔助內分泌治療、淋巴結陽性和HER2狀態)和9種蛋白(EGFR、MPHOSPH10、ACOX2、CASP3、ARL3、KRT18、FAM102A、STEAP3和BUB1B)。
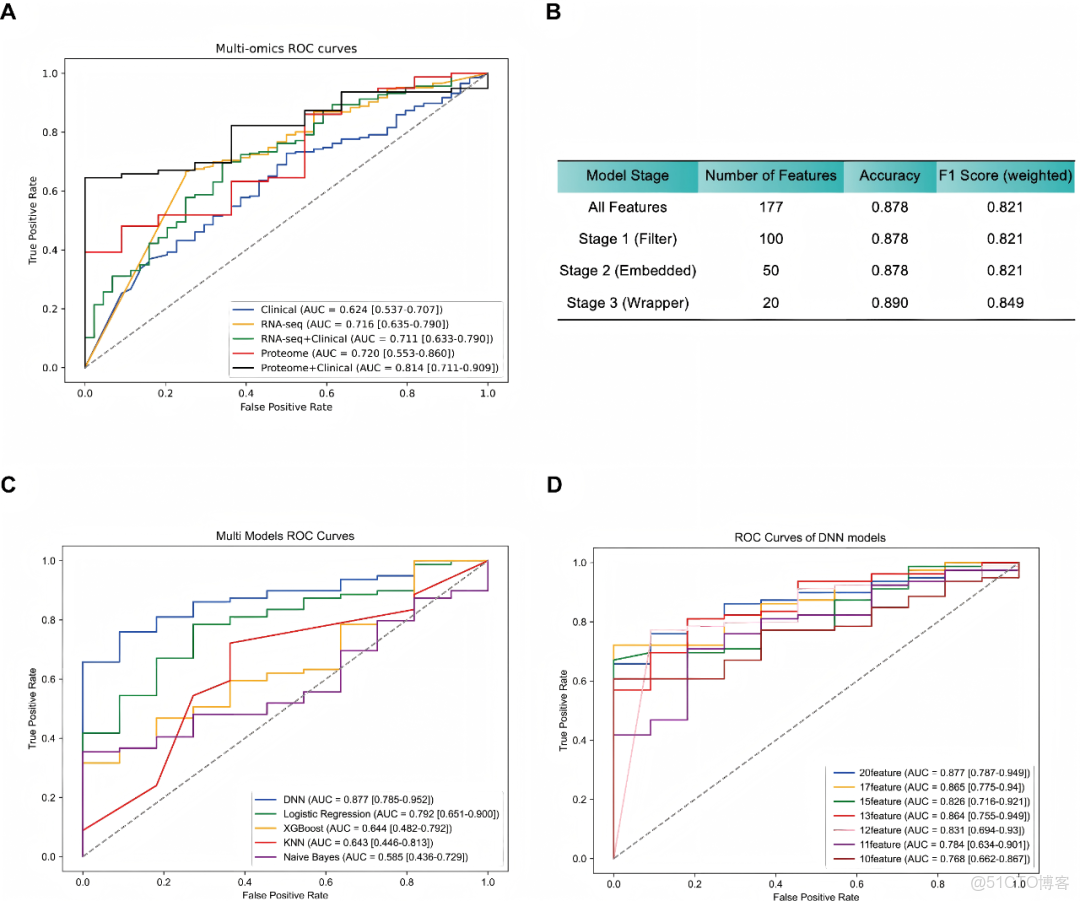
圖2. 預測模型的受試者工作特徵(ROC)曲線
(A)測試中五種不同數據組合的AUC。(B)三步特徵篩選策略。(C)五種機器學習模型的ROC曲線。(D)DNN模型的ROC曲線。
SHAP解釋
為闡明我們最終模型的有效性,我們採用SHAP方法來探究各特徵的貢獻度。環形圖和蜂羣圖顯示,對預測結果具有顯著貢獻的特徵包括MPHOSPH10、EGFR、ARL3、KRT18、淋巴結陽性、Her2狀態(免疫組化)、輔助內分泌治療、FAM102A、STEAP3、CASP3、腫瘤大小、BUB1B和ACOX2(圖3A,B)。圖3C展示了各特徵對預測結果的影響程度。MPHOSPH10、EGFR、ARL3等蛋白以及“淋巴結陽性”和“HER2狀態(免疫組化)”等特定臨牀特徵,通過其SHAP值在不同樣本間的顯著變異性和相對分散的顏色分佈,顯示出對模型結果的顯著影響。對於特定樣本,紅色標註的MPHOSPH10和EGFR高表達可能導致模型預測值升高。另一方面,相關性熱圖(圖3D)呈現了各特徵間關係的強度與方向。例如,“輔助內分泌治療”與自身的相關係數為1.00,而與“淋巴結陽性”的相關係數為0.19,呈弱正相關,與EGFR的相關係數為-0.36,呈中度負相關,與MPHOSPH10的相關係數為-0.22,呈弱負相關。此外,腫瘤大小與MPHOSPH10的散點圖展示於補充資料:圖2中。
此外,SHAP局部解釋闡明瞭各特徵如何影響每位患者5年生存預測(是或否)的概率。圖4A和4B分別展示了13個特徵的SHAP值決策過程,針對特定乳腺癌患者推演出無5年生存率(0.0)與5年生存率(1.0)的結果。圖4C和D的力圖揭示了不同特徵如何共同影響最終預測結果:當f(x)=0時,該乳腺癌患者的預測結果為無5年生存;當f(x)=1時,則預測結果為5年生存。
為評估模型在不同分子亞型間的穩健性,研究對Luminal A、Luminal B、HER2富集型和三陰性乳腺癌(TNBC)進行了分層分析。該模型取得了一致的高預測性能,各亞型曲線下面積(AUC)分別為:Luminal A型1.00、Luminal B型0.98、HER2富集型0.96、TNBC型0.92(補充資料:圖3A-D)。SHAP分析進一步揭示了各亞型內部特徵重要性的差異化模式:在Luminal A型中,輔助內分泌治療、MPHOSPH10和EGFR是最具影響力的預測因子;Luminal B型則以淋巴結陽性、ARL3和MPHOSPH10為主導;對於HER2富集型,淋巴結陽性、EGFR和MPHOSPH10排名最高;而在TNBC亞型中,MPHOSPH10、ARL3和EGFR的貢獻度最為顯著。這些發現證明該模型不僅能保持跨異質性亞型的卓越預測能力,還能精準捕捉各亞型特有的預後驅動因素。
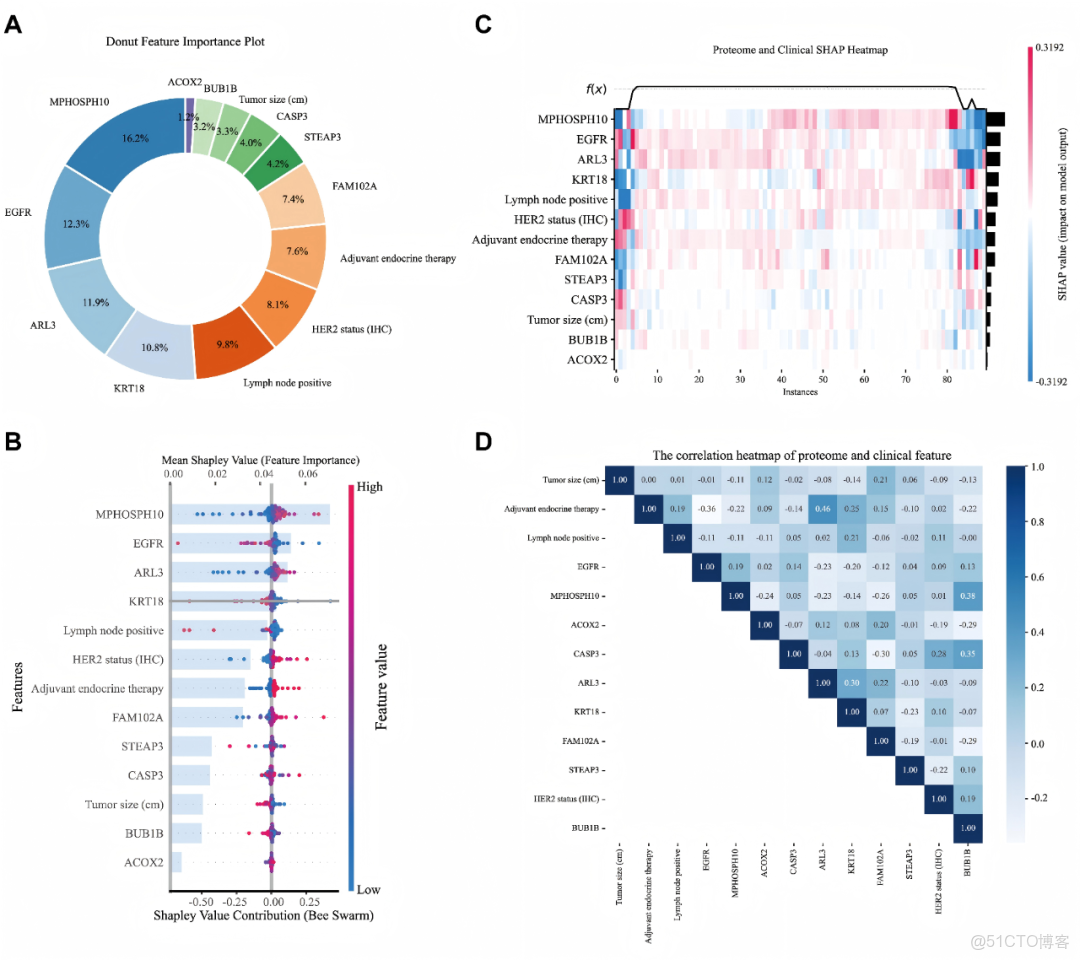
圖3. 使用SHAP進行全局解釋
(A)特徵重要性環形圖與(B)蜂羣圖。(C)熱力圖可視化。(D)13個特徵間的相關性熱力圖。
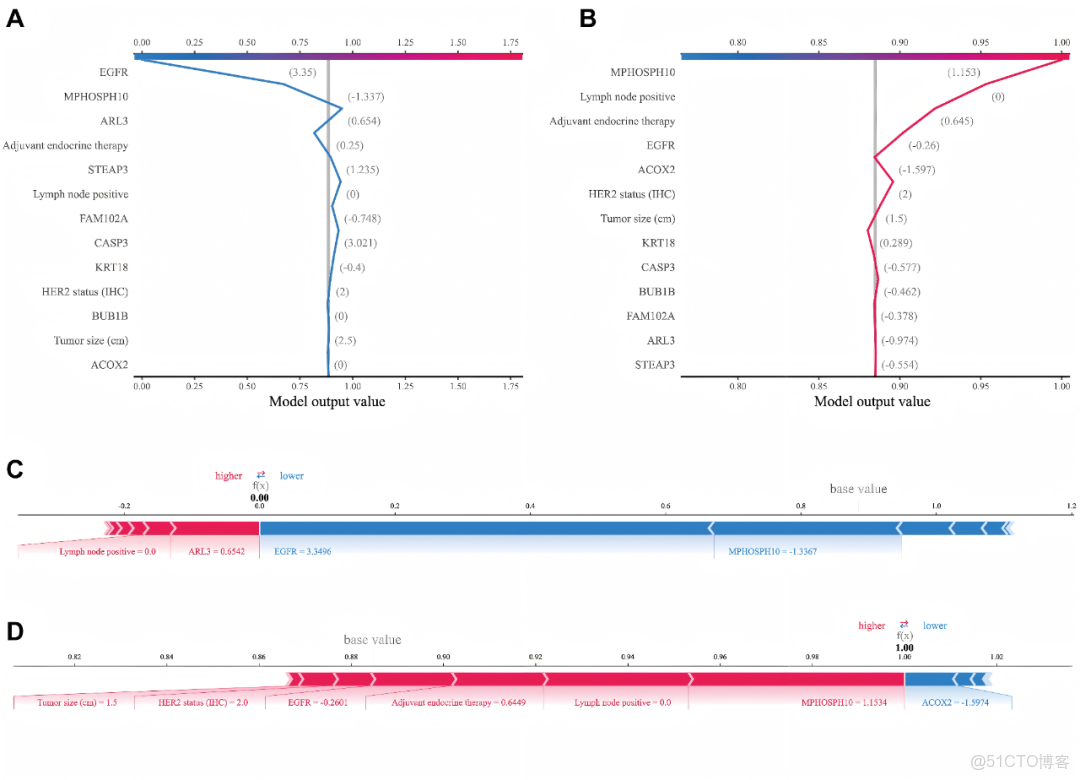
圖4. SHAP的局部解釋
(A,B)個體乳腺癌樣本的決策映射圖顯示:當f(x)為0時表示生存期不足5年,當f(x)為1時表示生存期超過5年。(C,D)該樣本預測結果的力圖。當f(x)為0.0時預測結果為生存期不足5年,當f(x)為1.0時預測結果為生存期超過5年。
KAN解釋與優化
為進一步提升最優模型的透明度和可解釋性,研究團隊採用KAN方法對整合蛋白質組學和臨牀特徵的13個特徵模型進行了驗證與解釋。受試者工作特徵(ROC)曲線分析顯示,AUC值達0.81(圖5A),具有良好的分類性能,表明其在乳腺癌5年生存預測中具有較強的判別能力。如圖5B所示,KAN網絡拓撲結構被詳細可視化,清晰闡釋了整合蛋白質標誌物(EGFR、MPHOS10、ACOX2、CASP3、ARL3、KRT18、FAM102A、STEAP3、BUB1B)與臨牀特徵(包括腫瘤大小、淋巴結轉移等腫瘤特性,以及內分泌治療等治療因素)的乳腺癌5年生存預測模型輸出結果。此外,擬合函數分析表明MPHOSPH10(R²=0.92)和腫瘤大小(R²=0.95)是模型預測結果的關鍵貢獻因子(圖5C,D),這些關鍵特徵與預測結果呈現顯著的線性相關性。通過KAN方法,我們成功量化了各特徵-結局關係的函數形式,並識別出具有強線性主導效應的特徵(如MPHOSPH10、腫瘤大小)。這種機制層面的可解釋性難以通過LIME等局部替代模型實現,因其無法直接表徵全局特徵-結局映射關係。這些發現有力驗證了KAN在解析生物醫學數據複雜相互作用方面的強大能力。
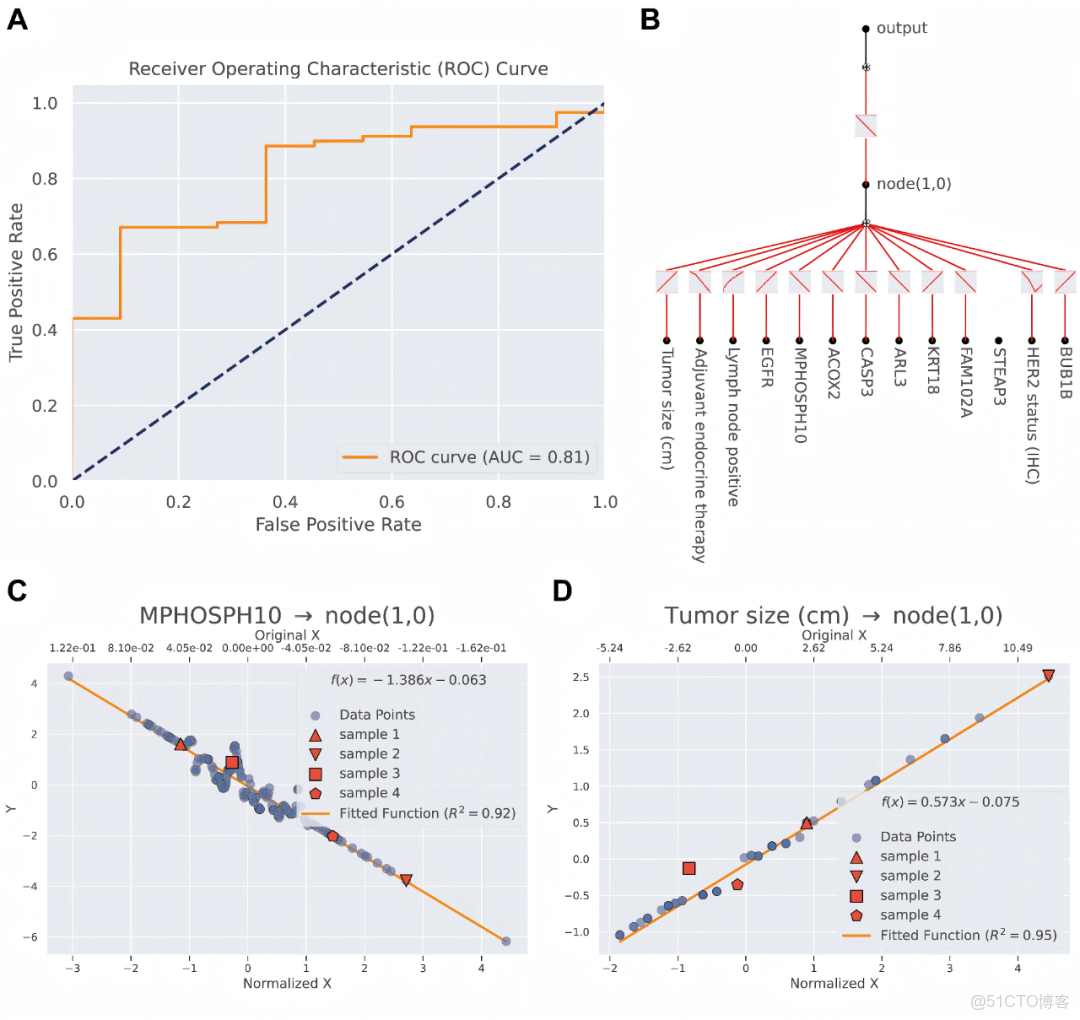
圖5. KAN解釋
(A)受試者工作特徵曲線。(B)網絡分析。(C)MPHOSPH10的線性擬合函數。(D)腫瘤大小的線性擬合函數。KAN,科爾莫戈洛夫-阿諾爾德網絡;ROC,受試者工作特徵曲線。
在線預測工具
為提升最終模型的臨牀應用價值,我們基於Streamlit Python框架開發了直觀的網絡應用程序(圖6),實現了乳腺癌5年生存預測模型的可視化部署。臨牀醫生或患者可通過左側交互界面輸入13項關鍵特徵值,右側SHAP力圖可實時評估5年生存預測結果,直觀展示各特徵對預測的貢獻度。該在線預測工具不僅支持快速臨牀決策,更通過SHAP解釋增強了臨牀醫師對模型的信任度。應用程序可通過以下鏈接訪問(https://ai-model-jhwvgzhyqyimdbvhptcxrp.streamlit.app/)。
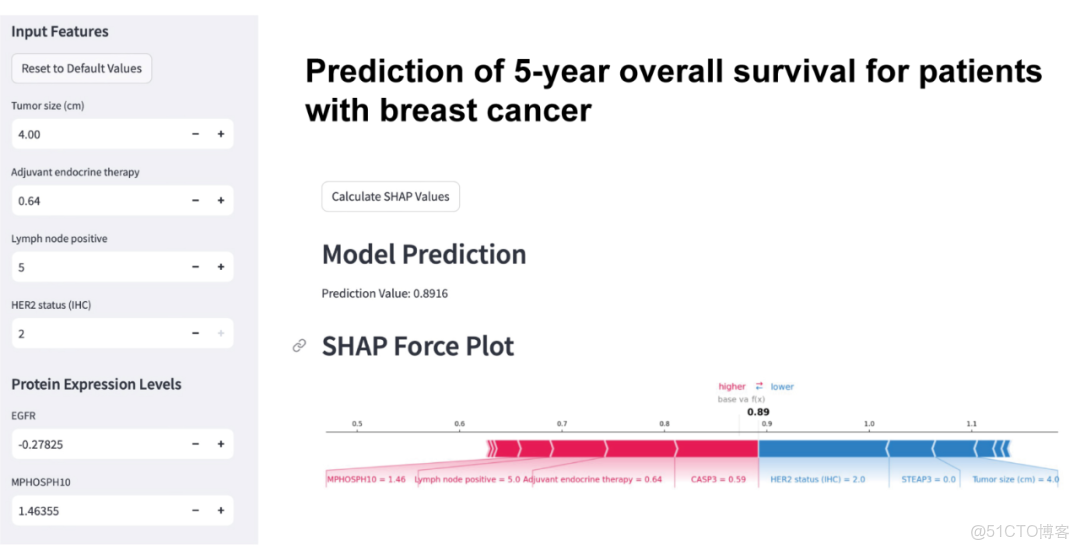
圖6. 乳腺癌5年生存期預測最優模型的在線預測平台,包含13個關鍵特徵
關鍵靶標驗證
最後,我們通過HPA數據庫研究了預後預測模型中9種蛋白質的表達情況,如圖7A-G所示。免疫組織化學染色分析顯示,與正常組織相比,MPHOSPH10、EGFR、ARL3、KRT18、STEAP3、CASP3和ACOX2在乳腺癌組織中存在差異表達。此外,關鍵蛋白的總生存分析還通過kaplan-Meier繪圖工具利用GEO數據庫進行了外部驗證,詳見補充資料:圖4。隨後,我們對乳腺癌組織進行了RNA測序以闡明關鍵靶點的表達水平。分析顯示,與癌旁組織相比,BUB1B和EGFR在乳腺癌組織中顯著上調,而ACOX2的表達在癌組織中明顯下調(補充資料:圖5)。
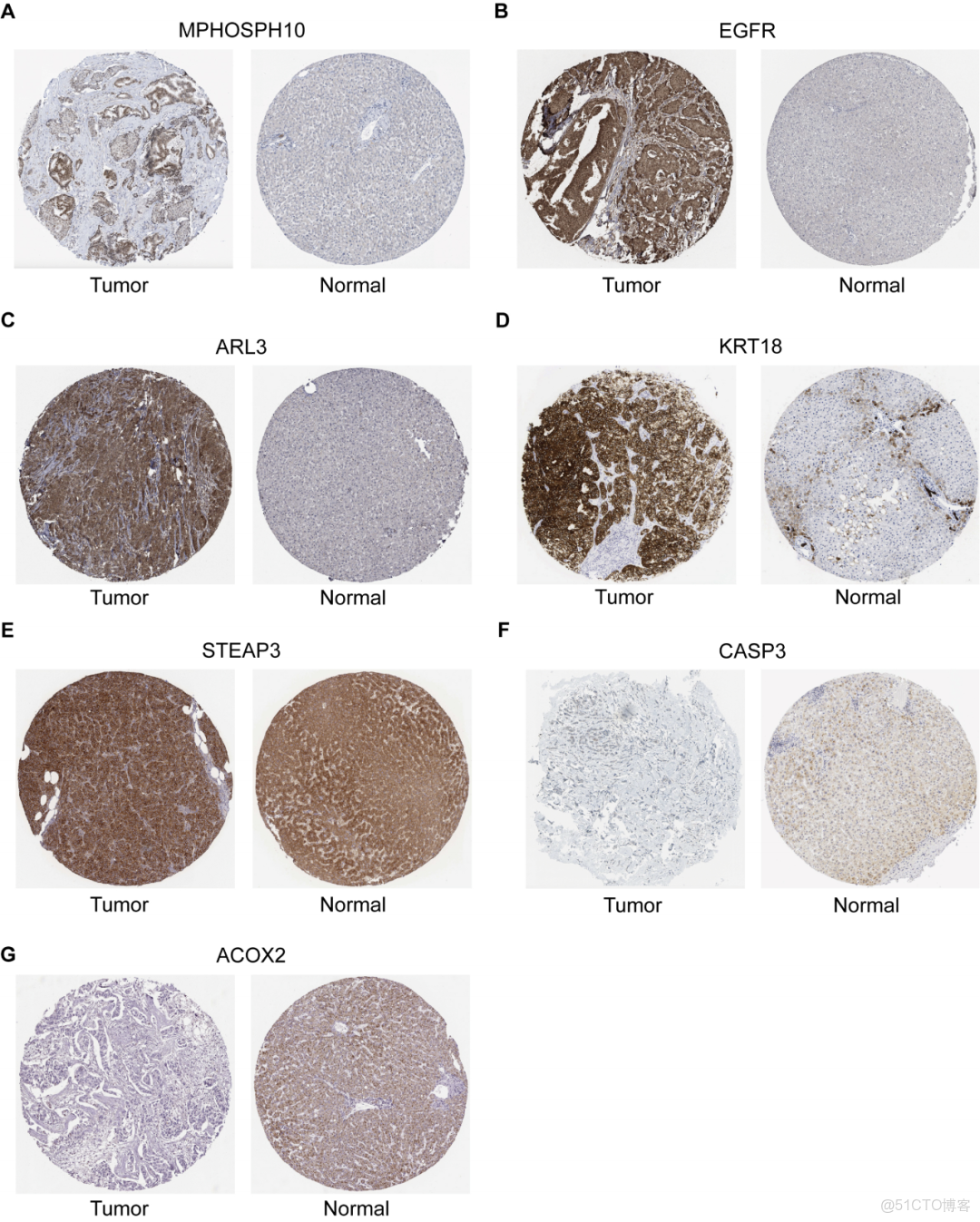
圖7. 通過HPA數據庫對預測預後模型關鍵蛋白進行免疫組化驗證
(A) MPHOSPH10. (B) EGFR. (C) ARL3. (D) KRT18. (E) STEAP3. (F) CASP3. (G) ACOX2.
討 論
本研究開發並驗證了一個整合蛋白質組學與臨牀數據的可解釋模型,用於預測乳腺癌患者5年生存率。研究結果表明,採用DNN算法將蛋白質組學特徵與既定臨牀參數相結合,其預測性能顯著優於僅基於臨牀特徵、RNA測序數據或蛋白質組學的模型。這個包含13個關鍵特徵的優化模型展現出卓越的判別能力,其AUC值達0.8642,精確率、召回率和F1分數均平衡在0.9367。通過SHAP和KAN方法的後續應用,我們深入理解了特徵貢獻度與交互作用,從而增強了模型的可解釋性。值得注意的是,本研究的最終預測模型雖然僅採用少量關鍵特徵,卻實現了優異性能。這主要歸因於以下因素:首先,通過嚴格的特徵選擇方法和可解釋性分析,我們精準篩選出最具信息量和相關性的變量;其次,降低特徵維度有助於緩解過擬合問題,提升模型泛化能力——這在樣本量有限時尤為重要;最後,採用的深度學習模型能夠捕捉選定特徵間複雜的非線性關係,即便輸入變量有限仍能進一步提高預測準確性。這些因素共同作用,使得精簡的特徵集也能產生穩健的預測性能。
多模態數據整合已被日益視為捕捉癌症進展和治療反應背後複雜生物過程的關鍵。值得注意的是,我們的研究結果表明,基於蛋白質組學的模型在預測臨牀結局方面優於基於RNA測序的模型。這種差異可能源於蛋白質組學直接測量蛋白質丰度,相比RNA表達水平能更準確地反映細胞功能狀態和生物通路活性。雖然RNA測序能提供基因表達的重要信息,但轉錄後修飾、蛋白質降解和其他調控機制可能導致mRNA與蛋白質水平不一致。因此,蛋白質組學數據能捕捉RNA測序單獨分析可能遺漏的其他生物複雜性層面,這解釋了其在本研究中更具預測優勢的原因。未來工作中整合這兩種數據類型有望進一步提升模型性能和生物學洞察力。
研究確定的13項特徵標誌物組合包含4項臨牀常規參數(腫瘤大小、輔助內分泌治療、淋巴結狀態和HER2狀態)及9種蛋白質標記物(EGFR、MPHOSPH10、ACOX2、CASP3、ARL3、KRT18、FAM102A、STEAP3和BUB1B)。通過SHAP分析,我們發現MPHOSPH10、EGFR、ARL3、KRT18、淋巴結狀態和HER2狀態是驅動預測結果最具影響力的特徵因子。這些發現與現有關於乳腺癌生物學特性及預後的認知相吻合。
在我們的KAN分析中,MPHOSPH10和腫瘤大小顯示出特別重要的預測價值,其R²值分別達到0.92和0.95。MPHOSPH10(M期磷蛋白10)參與核糖體生物合成和細胞週期進程,這些過程對癌細胞增殖至關重要。近期一項研究將MPHOSPH10鑑定為乳腺癌預後分層和治療指導的RNA結合蛋白之一,進一步證實了其在乳腺癌中的重要性。基於我們的研究發現,MPHOSPH10可能作為一種新型預後標誌物。同樣,其他幾種已鑑定的蛋白質,包括ARL3、STEAP3和FAM102A,也代表着值得在乳腺癌中進一步研究的潛在新型生物標誌物。我們模型中識別的這些蛋白質(如STEAP3)目前尚未納入常規臨牀檢測。由於大多數診斷實驗室尚未建立標準化且具有成本效益的檢測方法,這給直接臨牀轉化帶來了挑戰。
然而,靶向蛋白質組學(如多重反應監測)和多重免疫分析技術的進步,為開發這些生物標誌物的臨牀應用檢測提供了可行路徑。此外,將這些蛋白整合至現有的多標誌物組合或配套診斷試劑盒中,可促進其融入臨牀工作流程。在廣泛應用前,必須通過前瞻性研究評估這些檢測方法的可行性、可重複性及成本效益。
表皮生長因子受體(EGFR)過表達此前已被證實與乳腺癌不良預後相關,尤其在三陰性和炎性亞型中表現顯著。細胞骨架蛋白角蛋白18(KRT18)參與上皮-間質轉化及腫瘤進展過程。而凋亡執行關鍵蛋白半胱天冬酶-3(CASP3)的功能失調可能導致治療耐藥性。
應用先進的可解釋性技術(包括SHAP和KAN)通過清晰展示特徵對預測結果的貢獻度,既增強了臨牀醫生的信任度,又促進了對疾病機制的生物學理解。輔助內分泌治療與EGFR之間中等程度的負相關性(-0.36)符合已知的內分泌耐藥機制——EGFR通路激活可促進雌激素非依賴性生長。
儘管KAN與SHAP的整合相較於傳統深度神經網絡顯著提升了模型透明度,但這些方法仍無法完全消除深度學習的“黑箱”特性。SHAP能從全局和局部兩個維度量化每個特徵對預測結果的貢獻度,而KAN則能可視化已學習到的函數關係,從而提供有價值的解釋性見解。然而,這兩種方法均屬於事後可解釋性工具,既不能確保完全的因果性,也無法提供機制性理解。因此,在臨牀決策應用中,模型預測結果必須結合生物學知識進行解讀,並通過實驗研究加以驗證。
SHAP與KAN分析結果之間存在明顯差異。SHAP通過計算每個特徵在所有樣本中對模型預測的平均邊際貢獻來分配重要性分數,反映的是特徵的全局影響力。而KAN則量化每個特徵與結果之間關係的強度及線性/非線性特性,這種方法可能凸顯出在特定患者亞羣中具有強直接效應的特徵,即使這些特徵的整體全局影響較低。例如,腫瘤大小在KAN分析(R²=0.95)中表現出高度線性主導性,但在SHAP排名較低,這可能説明其在特定臨牀情境下具有顯著預後相關性,而在整個隊列中的平均貢獻相對較小。這種互補性視角凸顯了採用多種可解釋性技術來更全面理解模型行為的重要價值。
開發基於網絡的可訪問預測工具標誌着我們模型向臨牀轉化邁出了重要一步。通過SHAP力圖提供直觀的特徵貢獻可視化,臨牀醫生能更好地理解個體患者預測依據,有望輔助治療決策和風險分層。
必須承認我們的研究存在一些侷限性。本研究的一個侷限在於,除用於外部驗證的公開數據集外,缺乏其他前瞻性真實世界臨牀隊列數據。雖然現有隊列提供了多樣化的患者羣體並增強了研究結果的普適性,但可能無法全面反映更廣泛臨牀環境中乳腺癌的異質性。未來工作將開展多中心前瞻性數據收集,以在實際臨牀環境中進一步驗證和完善該模型。其次,當前分析將5年生存率作為二分類結局指標,後續研究應考慮採用時間-事件分析和競爭風險模型。
結 論
我們結合蛋白質組學和臨牀數據的可解釋多模態模型在預測乳腺癌患者5年生存率方面展現出穩健性能。通過SHAP和KAN技術揭示的蛋白質標誌物,尤其是MPHOSPH10,展現出作為預後生物標誌物和治療靶點的良好潛力,值得深入研究。先進可解釋性技術的整合與便捷預測工具的研發,提升了該模型臨牀轉化的可能性,有力推動了精準腫瘤學在乳腺癌治療領域的應用前景。
方 法
研究人羣與數據來源
從復旦大學附屬腫瘤醫院( Fudan University Shanghai Cancer Center,FUSCC )靶向測序隊列、TCGA研究網絡、乳腺癌分子分類國際聯盟和臨牀蛋白質組學腫瘤分析聯盟獲取乳腺癌的轉錄組數據、蛋白質數據和臨牀數據。這項研究包括了來自中國各地的773名未經治療的乳腺癌患者,他們在2013年和2014年在FUSCC接受了治療。這些患者術前均未接受過系統治療(包括化療、內分泌治療、靶向治療或免疫治療等)或放療。用於Kaplan - Meier驗證的GEO數據集包括:E‐MTAB‐365 (n = 537), E‐TABM‐43 (n = 37), GSE11121 (n = 200), GSE12093 (n = 136), GSE12276 (n = 204), GSE1456 (n = 159), GSE16391 (n = 55), GSE16446 (n = 120), GSE16716 (n = 47), GSE17705 (n = 196), GSE17907 (n = 54), GSE18728 (n = 61), GSE19615 (n = 115), GSE20194 (n = 45), GSE20271 (n = 96), GSE2034 (n = 286), and GSE20685 (n = 327), GSE20711 (n = 90), GSE21653 (n = 240), GSE22093 (n = 68), GSE25066 (n = 507), GSE26971 (n = 276), GSE29044 (n = 79), GSE2990 (n = 102), GSE31448 (n = 71), GSE31519 (n = 67), GSE32646 (n = 115), GSE3494 (n = 251), GSE36771 (n = 107), GSE37946 (n = 41), GSE41998 (n = 279), GSE42568 (n = 121), GSE43358 (n = 57), GSE43365 (n = 111), GSE45255 (n = 139), GSE4611 (n = 153), GSE46184 (n = 74), GSE48390 (n = 81), GSE50948 (n = 156), GSE5327 (n = 58), GSE58812 (n = 107), GSE61304 (n = 62), GSE65194 (n = 164), GSE6532 (n = 82), GSE69031 (n = 130), GSE7390 (n = 198), GSE76275 (n = 265), GSE78958 (n = 424), GSE9195 (n = 77).
數據處理
我們共獲得773例27個臨牀特徵(附表1),752例211個特徵(補充資料:表2和表3)的RNA測序數據,271例151個特徵(補充資料:表4和表5)的蛋白質組學數據。從FUSCC電子病歷中檢索臨牀信息,包括診斷年齡、腫瘤大小、組織學分級、淋巴結狀態、雌激素受體( ER )狀態、孕激素受體(PR )狀態、HER2狀態和生存結果。當缺失率為5 %時,缺失的臨牀值被排除在分析之外,使用鏈式方程多重填補。基因表達值在分位數標準化後被標準化為每百萬個log2轉化的轉錄本。蛋白質丰度進行log2‐轉換,並使用跨樣本中位數中心化進行標準化。在低於80 %的樣本中檢測到的蛋白質被排除,缺失值用基於正態分佈的方法(寬度= 0.3 ,下移= 1.8 )填補。蛋白組學數據和轉錄組學數據兩者之間有77個交點,如補充資料所示:表6。主要研究終點為5年總生存期( OS ),定義為從確診之日起至60個月內任何原因死亡的時間。存活超過60個月的患者視為存活患者,而在60個月之內失訪的患者在最後一次接觸的日期進行刪失。為了確定最優的多組學特徵,將具有不同特徵的數據進行如下劃分:a .臨牀特徵b . RNA測序數據c .蛋白質組學數據d .臨牀特徵結合RNA測序數據。e.臨牀特徵結合蛋白質組學數據。5年以上生存定義為1,5年以下生存定義為0。對於基於轉錄組學的多組學模型,將臨牀特徵與來自同一患者的轉錄組圖譜進行整合,形成輸入特徵矩陣。對於基於蛋白質組學的多組學模型,臨牀特徵與相應患者的蛋白質組圖譜進行了整合。在每種情況下,僅將兩種模態數據均完整的樣本納入各自的分析。在整合之前,所有特徵在每個模態內使用z‐score變換進行獨立標準化,以確保跨數據類型的可比性。
模型開發與比較
將整合後的表達數據隨機分為訓練集( 70 % )和測試集( 30 % )。訓練集用於擬合模型參數和調整超參數,測試集用於評估預測性能和泛化能力。我們考察了訓練隊列中存活和非存活病例的分佈,發現中等程度的類別不平衡。為了減輕模型訓練過程中的潛在偏差,在損失函數中應用了與類頻率成反比的類權重。這種方法確保了在沒有對數據進行過採樣或欠採樣的情況下,兩類對優化過程的貢獻相等。採用深度神經網絡( DNN )、邏輯迴歸( LR )、極端梯度提升( XGBoost )、K近鄰算法( KNN )和樸素貝葉斯法( NB )五種機器學習算法構建乳腺癌預後預測模型。對於所有模型,在訓練集上進行5折交叉驗證,以防止過擬合。具體來説,使用交叉熵作為損失函數的Adam優化器來訓練DNN模型。採用如下超參數進行訓練:學習率:0.001,批量大小:32,訓練輪數:100,激活函數:ReLU用於隱藏層、sigmoid用於輸出層,刪除比率:0.3,以此來減輕過擬合。為了提高模型的泛化能力和防止過擬合,我們採用了提前停止的方法,如果連續10輪訓練的驗證損失沒有減少,則停止訓練。保留表現最好的模型(基於驗證損失)進行下游評價。
模型評價
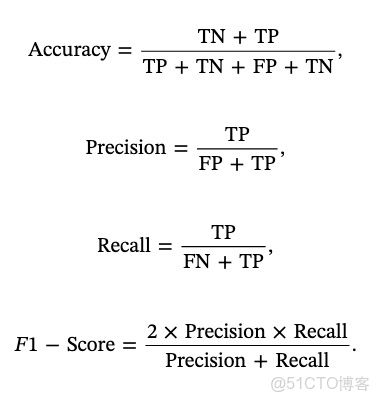
為了評估模型的整體性能,測量了受試者工作特徵曲線下的總面積( AUC )、準確率、精確率、召回率和F1分數。計算公式如下:Tp:真陽性;Tn:真陰性;Fp:假陽性;Fn:假陰性。
最優特徵選擇
採用三步特徵篩選策略來確定最具信息量的變量。初始的177個特徵首先根據統計顯著性被過濾為100個候選特徵。然後使用嵌入的方法將該集合減少到50個特徵。最後,採用基於包裝器的方法選擇具有最大預測相關性(補充資料:表7)的前20個特徵。使用深度DNN構建了初步模型。在訓練過程中,應用Adam優化器,以交叉熵作為損失函數。為了識別最重要的特徵,我們使用SHAP來評估特徵對訓練好的DNN模型的貢獻。SHAP為每個特徵分配一個Shapley值,代表其對模型輸出的邊際貢獻,允許我們根據特徵的平均絕對SHAP值對特徵進行排序。基於此排名,我們選取了排名前20、前17、前15、前13、12、11、10的特徵進行下游模型構建和對比。
模型解釋
SHAP
為了闡明具有“黑箱”特徵的ML模型的內在機制,我們系統地使用SHAP框架來解釋最優預測模型。這種基於博弈論的方法在現有研究中得到了廣泛的驗證,它通過Shapley值分解提供了有數學基礎的解釋。通過彙總圖實現全局解釋,可視化整體特徵重要性,量化輸入變量和輸出預測之間的方向關係。對於局部解釋,力圖通過顯式地顯示每個特徵值如何增加或減少特定實例的基線預測概率來分解個體預測。SHAP值是通過Shapley值分解計算得到的,它量化了每個特徵對模型預測的邊際貢獻,同時考慮了所有可能的特徵交互。
KAN
基於科爾莫戈洛夫-阿諾爾德表示定理的KAN算法也提高了乳腺癌5年生存率預測模型的性能。網絡和函數表達式為輸入變量和預測輸出之間提供了數學基礎。其規範化形式如下:
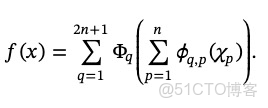
為了可視化內部架構並解釋學習到的表徵,生成了網絡拓撲圖,顯示隱藏節點的組織及其激活模式。對於每個選定的特徵,直接從訓練好的KAN中提取相應的函數映射,並將其表示為顯式的數學公式( e.g. , f ( x) = a + bx ),通過R2統計量擬合曲線圖來進一步説明,以反映近似質量。通過計算關鍵節點的局部激活值,KAN提供了在每個樣本的基礎上最強烈地促進或抑制疾病分類的基因。這使得在羣體和個體水平上都有生物學意義的解釋成為可能。
關鍵靶標表達的驗證
收集温州醫科大學附屬第一醫院26例乳腺癌組織及癌旁組織。使用TRIzol試劑( TaKaRa )進行總RNA提取。使用Agilent 2100生物分析儀評估RNA樣品的完整性,隨後在Illumina平台上進行RNA測序。在人類蛋白質圖譜(https://www. proteinatlas.org/)中驗證乳腺癌與正常之間預後預測模型中關鍵蛋白的蛋白表達情況。
統計學分析
本研究採用R (版本 4.4.1)和Python (版本 3.9.12 )進行統計分析。所有統計評估均採用雙側檢驗,p值小於0.05表明差異有統計學意義。無序分類資料組間比較採用Pearson ' s χ 2檢驗或Fisher確切概率法,有序分類資料組間比較採用Mann - Whitney U檢驗。使用Kaplan - Meier繪圖儀對關鍵蛋白的總體生存分析進行外部驗證。
代碼和數據可用性:
本研究所用的轉錄組和蛋白質組數據均可在https://data.3steps.cn/cdataportal/網站獲得。完整的分析代碼可在https://github.com/2729956566/streamlit_ breast上通過GitHub訪問。
引文格式:
Wu, Z., Yao, S., Jin, L., Wu, X., Zhang, R., Wang, O. and Xia, E. (2025), An Interpretable Machine Learning Model for Predicting 5-Year Survival in Breast Cancer Based on Integration of Proteomics and Clinical Data.iMetaMed e700010. https://doi.org/10.1002/imm3.700010.
作者簡介

吳志炫(第一作者)
● 浙江大學在讀博士研究生。
● 研究方向為腫瘤神經微環境及腫瘤生物標誌物研究,以第一作者在Phytomedicine、Molecular Medicine、Computers in biology and medicine、Frontiers in immunology、BMC Cancer、imetaMED等期刊發表SCI論文10餘篇。

夏二傑(通訊作者)
● 温州醫科大學附屬第一醫院乳腺外科主治醫師。
● 研究方向為小分子藥物抗腫瘤藥物作用及機制研究、腫瘤生物信息學分析及預測模型構建。主持浙江省自然科學基金1項和温州市科技局課題1項。以一作和通訊作者在Phytomedicine、Computers in biology and medicine、International journal of oncology、imetaMED等高影響力期刊上發表研究論文。