
在結構生物學的發展歷史中,「結構決定功能」一度被視為近乎不可動搖的基本法則。無論是胰島素的經典螺旋構象,還是血紅蛋白的四聚體架構,都強化了一個共識:蛋白質要發揮生物學作用,必須擁有穩定的三維結構。
然而,內在無序蛋白(IDPs)及其內在無序區域(IDRs)的發現, 正不斷重塑這一傳統認知。它們在生理條件下並不形成固定結構,卻深度參與信號轉導、基因轉錄調控等核心過程,並與癌症、神經退行性疾病等重大人類疾病密切相關。
計算生物學研究進一步揭示,真核生物蛋白質組中約 30% 的氨基酸殘基處於無序狀態。這意味着,無序並非「異常」,而是生命體系的常態組成部分。然而,無序蛋白的高度動態性使其難以用傳統實驗技術穩定捕捉,也難以通過常規計算方法準確模擬其構象分佈, 這也成為了該領域長期存在的技術瓶頸。
圍繞這一挑戰,英國蛋白質分析技術研發商 Peptone 公司、哥本哈根大學、英偉達、牛津大學、麻省理工學院、杜克大學等組成的聯合團隊,提出了兩項關鍵突破。其一是 PeptoneBench 系統評估框架, 該框架整合 SAXS、NMR、RDC、PRE 等多源實驗數據,並結合最大熵重加權等統計方法,實現了實驗觀測與理論預測的嚴格定量對照。其二是生成式模型 PepTron, 基於擴展後的合成 IDR 數據集訓練,專門強化了對無序區域的建模能力,使其能夠更好地捕捉無序蛋白的構象多樣性。
研究團隊利用 PeptoneBench 將 PepTron 與 AlphaFold2、Boltz2、BioEmu 等主流預測工具進行了系統對比,結果顯示,PepTron 在有序與無序區域的預測上均展現出與實驗結果高度一致的表現,達到了 SOTA 水平。基於這些進展,一個更準確、更貼近生物現實的「構象集合」結構預測框架正在形成,顯著提升了人們對蛋白質在有序—無序全譜狀態下的整體理解能力。
相關研究成果以「Advancing Protein Ensemble Predictions Across the Order–Disorder Continuum」為題,已發表預印本於 bioRxiv。

論文地址:
https://www.biorxiv.org/content/10.1101/2025.10.18.680935v1
關注公眾號,後台回覆「PepTron」獲取完整 PDF
更多 AI 前沿論文:
https://hyper.ai/papers
PeptoneBench 與多源實驗數據集的系統化構建
蛋白質數據庫(PDB)是結構生物學最基礎、最重要的公共資源,但在內在無序蛋白(IDPs)及其無序區域(IDRs)的覆蓋上卻存在明顯結構缺口——僅約 3% 的條目被標註為無序, 而在人類蛋白質組中,此類無序區域的比例卻高達 20–30%。
如下圖所示,這種系統性偏斜使得大多數結構預測模型天然「偏愛」穩定構象,對動態無序狀態的學習能力長期受限。為彌補這一不足,研究人員引入瞭如 IDRome 等補充性數據庫,其無序佔比高達約 77%, 可從統計分佈上與 PDB 形成互補。然而,該數據庫缺乏真實實驗解析的結構數據,因而難以用作建模與評估的直接基準,其應用價值仍受到明顯限制。
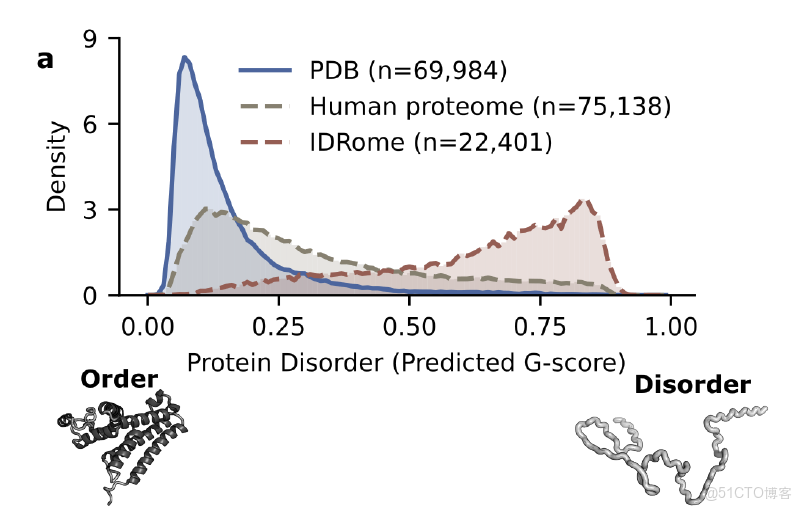
::: hljs-center
PDB、人類蛋白質組和 IDRome 數據集中預測的蛋白質無序分佈
:::
要突破上述數據瓶頸,第一步是建立可量化、可比較的無序程度指標。 該研究以蛋白質平均 G 評分為核心度量,其取值範圍介於 0(完全有序)到 1(完全無序),基於 NMR 二級化學位移(CS)數據計算,可準確反映局部二級結構形成的傾向。對於缺乏實驗 CS 數據的蛋白質,研究團隊採用基於 TriZOD 訓練的機器學習模型 ADOPT2 進行 G 評分預測,從而實現對整個有序–無序譜的統一量化。
在此基礎上,團隊進一步指出,僅依賴 PDB 的結構數據無法客觀評估構象集合的質量,因此必須構建覆蓋完整有序–無序範圍的實驗數據集。
為此,如下表所示,研究人員建立了 3 個互補的數據資源:PeptoneDB-CS(源於 BMRB 的 NMR 化學位移)、PeptoneDB-SAXS(來自 SASBDB 的 SAXS 圖譜)以及 PeptoneDB-Integrative(整合多種正交實驗數據的 IDP 專用集)。三類數據結構不同、信息互補,CS 揭示局部結構,SAXS 反映整體構象,Integrative 支持交叉驗證。
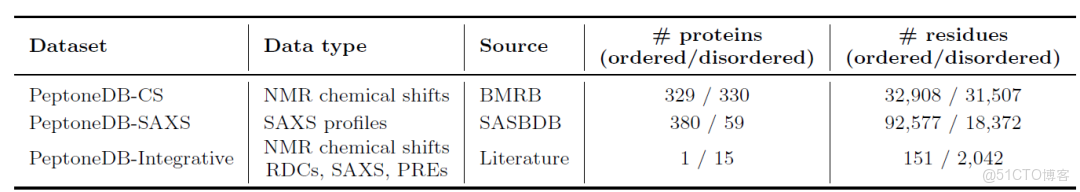
::: hljs-center
研究構建的蛋白質數據集
:::
基於這些數據,如下圖所示,研究人員構建了 PeptoneBench 評估框架,用於量化預測構象集合與實驗數據的一致性。 整個流程包括:構象集合標準化與預處理;通過正向模型將預測結構映射為可與實驗對比的觀測量;基於歸一化 RMSE 進行一致性評分,並在全程納入模型與實驗的不確定性。最終結果以 RMSE–G 評分關係圖展示,並通過 LOWESS 平滑與自舉法估計誤差,進一步綜合為 PeptoneBench 彙總評分,形成可直接比較不同工具性能的量化標準。
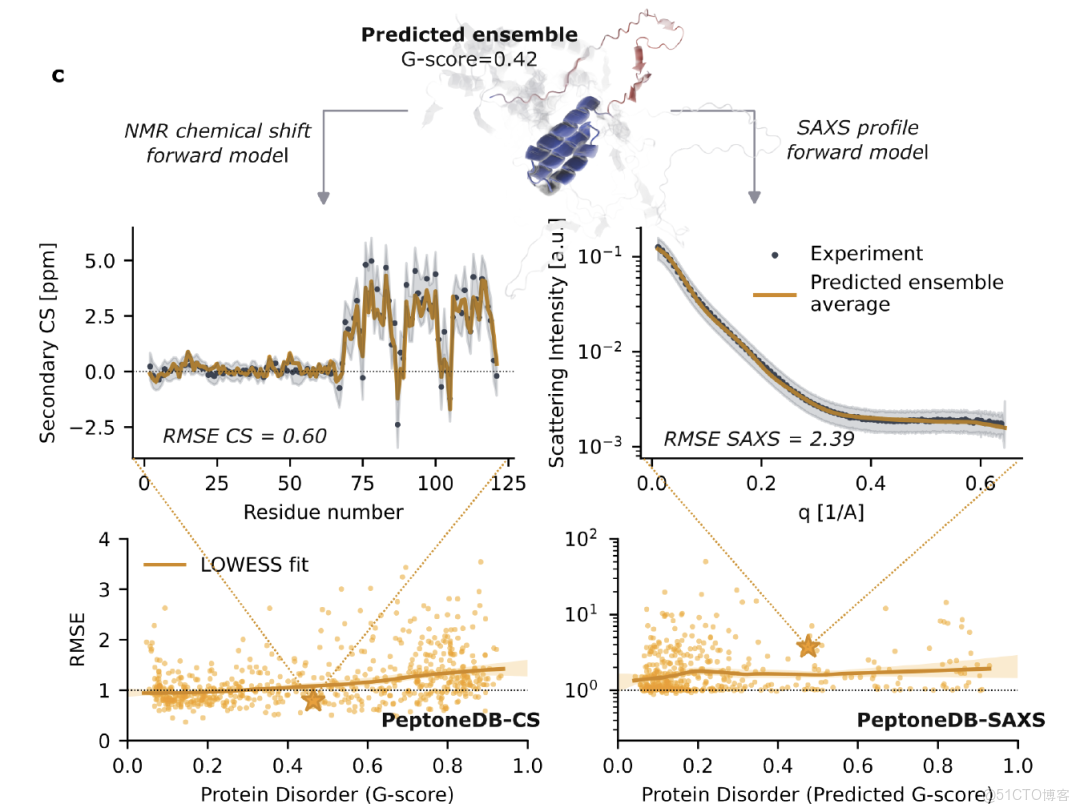
::: hljs-center
PeptoneBench 工作流程
:::
值得特別強調的是,某些初始 RMSE 較高的構象集合,通過最大熵重加權後反而可能更接近實驗分佈。為避免將「錯誤的權重」誤判為「缺失的構象」,PeptoneBench 同時報告重加權前後的 RMSE,以區分可修正的採樣偏差與不可恢復的構象缺失。 這一策略對高度動態、對實驗條件極其敏感的 IDP 尤為關鍵:只要生成模型能夠覆蓋足夠豐富的構象空間,即便實驗環境不同,也能通過重加權過程快速適配,從而顯著提升預測結果的實用性與可靠性。
PepTron:兼顧有序與無序蛋白的構象生成模型
該研究提出的 PepTron 模型,是一款基於流匹配 ESMFlow 架構構建的蛋白質構象生成器,目標是覆蓋從完全有序到高度無序的完整構象譜,生成既具物理合理性又具有結構多樣性的構象集合。
在模型架構上,PepTron 以 ESMFlow 為基礎,並在 NVIDIA BioNeMo 框架中實現以提升訓練與推理效率。 模型集成了 cuEquivariance 三角注意力機制,並通過 BioNeMo 的 Modular Co-Design 子包支持流匹配功能。訓練流程遵循 BioNeMo 的分佈式最佳實踐,結合多種並行策略和混合精度計算,因而能夠在多 GPU 環境中穩定、高效地擴展。
值得強調的是,PepTron 在推理階段不依賴多序列比對(MSA)或外部 ESM 權重,僅憑單一檢查點即可生成完整構象集合,大幅簡化了使用門檻。
針對無序區域實驗結構數據稀缺的難題,研究團隊基於 IDRome 構建了合成結構數據集 IDRome-o。為此,他們開發了基於片段組裝的蛋白質結構生成工具 IDP-o,能夠以極低成本大規模生成物理合理的 IDP 構象集合。 IDP-o 結合片段組裝與分層鏈增長策略,從包含 2.14 億結構的 AlphaFold 大型數據庫中提取六殘基片段,從而更準確地捕捉無序蛋白中短暫出現的螺旋結構。
需要説明的是,IDR-o 的目標並非模擬某一平衡分佈,而是覆蓋序列可能採樣到的所有合理構象,因此其輸出特別適用於後續的最大熵重加權,也可作為分子動力學模擬的高質量初始構象庫。
為克服傳統模型傾向預測穩定結構的偏差,如下圖所示,PepTron 採用了「實驗數據+合成數據」的混合訓練策略: 先使用 PDB 中的實驗解析結構進行預訓練,再引入合成生成的無序蛋白集合進行混合微調,從而讓模型充分學習有序與無序構象的連續分佈。即使在計算資源受限條件下,這種策略仍顯著提升了模型在各類蛋白上的預測性能。
在具體訓練流程上,研究分為兩個階段: 在基礎階段以 ESMFold 權重為起點,利用 PDB 數據對流匹配模塊進行再訓練,並將序列長度裁剪範圍擴展至 512 個殘基;在混合微調階段使用由 PDB 實驗結構與 IDRome-o 合成數據構成的混合集合作為訓練數據,對模型進行最終優化。這樣的設計使 PepTron 能夠打通有序—無序全譜,實現對蛋白質動態構象空間更全面、更真實的建模。
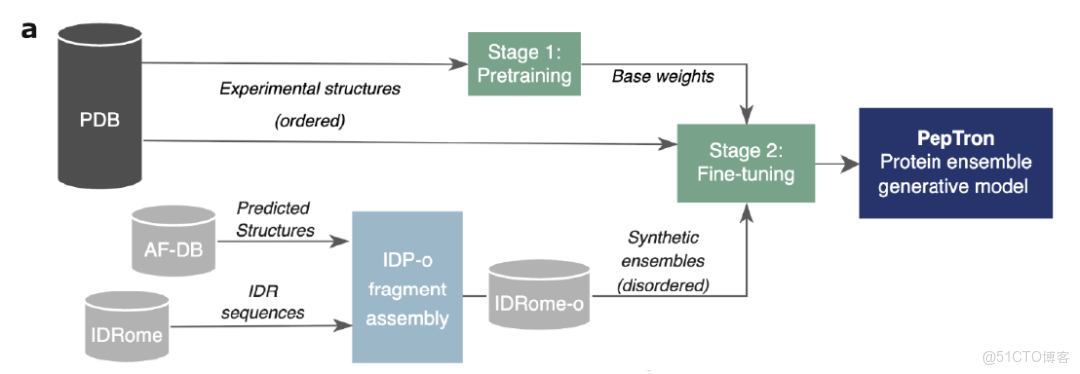
::: hljs-center
PepTron 模型示意圖
:::
面向全譜構象的模型驗證:PepTron 與主流方法的系統比較
研究團隊隨後使用 PeptoneBench 框架,在完全獨立於訓練集的實驗數據上系統評估 PepTron 的性能,並與 ESMFold、ESMFlow、AlphaFold2、Boltz2、BioEmu 等主流模型進行基準對比。同時,團隊在專注於內在無序蛋白(IDP)的 PeptoneDB-Integrative 數據集上開展專項測試,以全面檢驗各模型在無序構象建模上的能力。結果顯示出清晰的模型分化特徵。
如下圖所示,在 PeptoneDB-CS 數據集上,各模型的表現隨蛋白質無序程度(G 評分)呈現顯著差異:ESMFold 與 ESMFlow 在有序區域預測精確,但在無序區域性能明顯降低;IDP-o 則呈現典型的互補模式——無序度越高性能越好;而 PepTron 在整個有序—無序構象譜上都保持穩定的高一致性。 這種均衡能力在 PeptoneDB-SAXS 數據集以及隨後的重加權分析中再次被驗證,説明 PepTron 在不犧牲有序結構精度的前提下,能夠有效捕捉無序蛋白的構象多樣性。
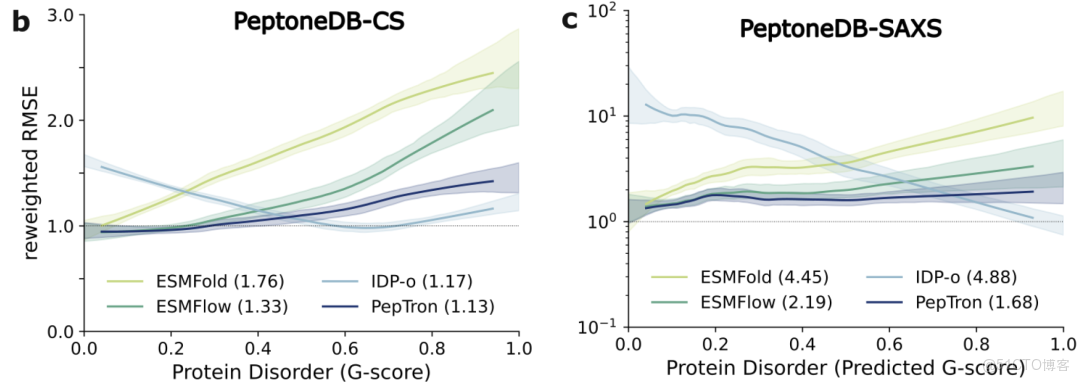
::: hljs-center
不同模型的 PeptoneDB-CS/SAXS 對比結果
:::
進一步的跨模型對比結果如下圖所示,AlphaFold2 與 Boltz2 雖然在有序蛋白的預測中依舊佔優,但隨着無序程度上升,其性能出現系統性衰減;相比之下,PepTron 與 BioEmu 在整個構象譜上都保持更強的穩健性,更適合處理 IDP 高度異質的結構特徵。
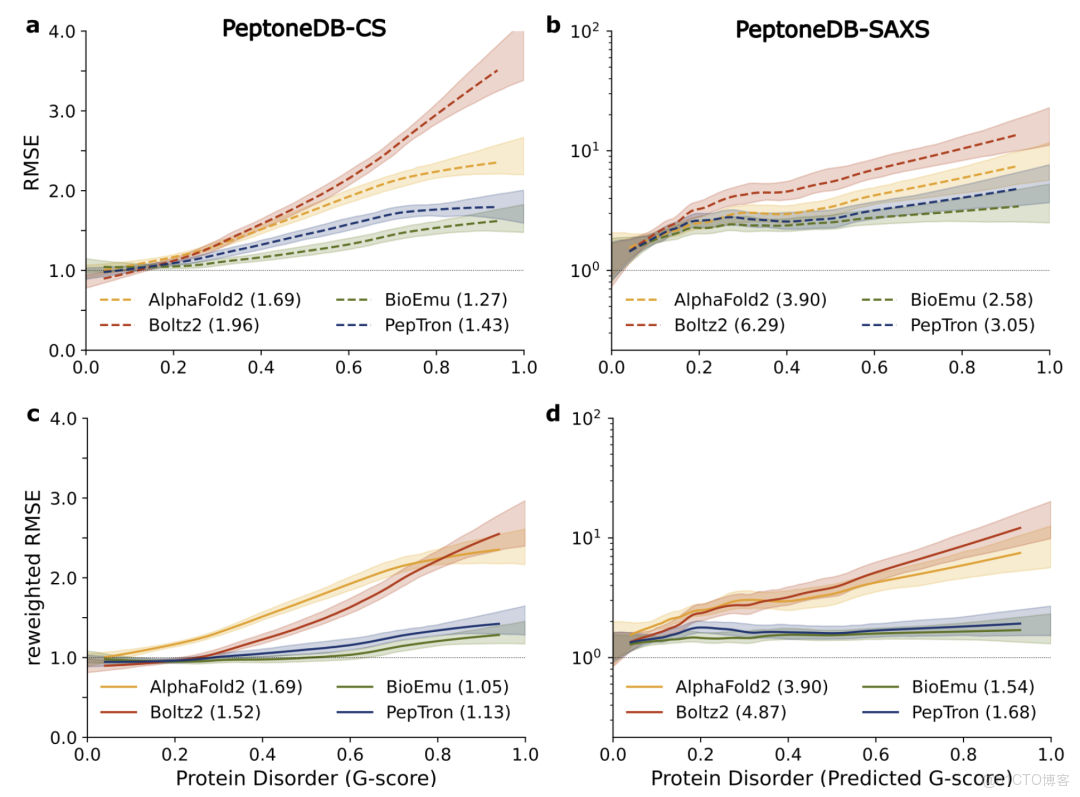
::: hljs-center
不同模型的 PeptoneDB-CS/SAXS 對比結果
:::
為了確保無序區域訓練不會損害其對有序蛋白的預測能力,研究團隊還在 CAMEO22 和 CASP14 的有序結構數據上進行了額外測試。結果表明,PepTron 在 RMSD、LDDT、TM 等關鍵指標上與 ESMFlow 表現一致,證明其在擴展 IDR 建模能力的同時並未削弱對有序結構的準確性。
在集成多實驗指標的 PeptoneDB-Integrative 數據集中,如下圖所示,模型表現進一步呈現差異。IDP-o 在最大熵重加權後表現尤為突出,無論是 RMSE 還是 RDC Q 因子均顯著優於其他模型;PepTron 與 BioEmu 在 RDC 指標上接近,但 BioEmu 在局部化學位移預測中更具優勢。值得注意的是,即便在未重加權條件下,IDP-o 仍在多數局部與全局指標中領先,體現其在無序蛋白構象覆蓋上的天然優勢。
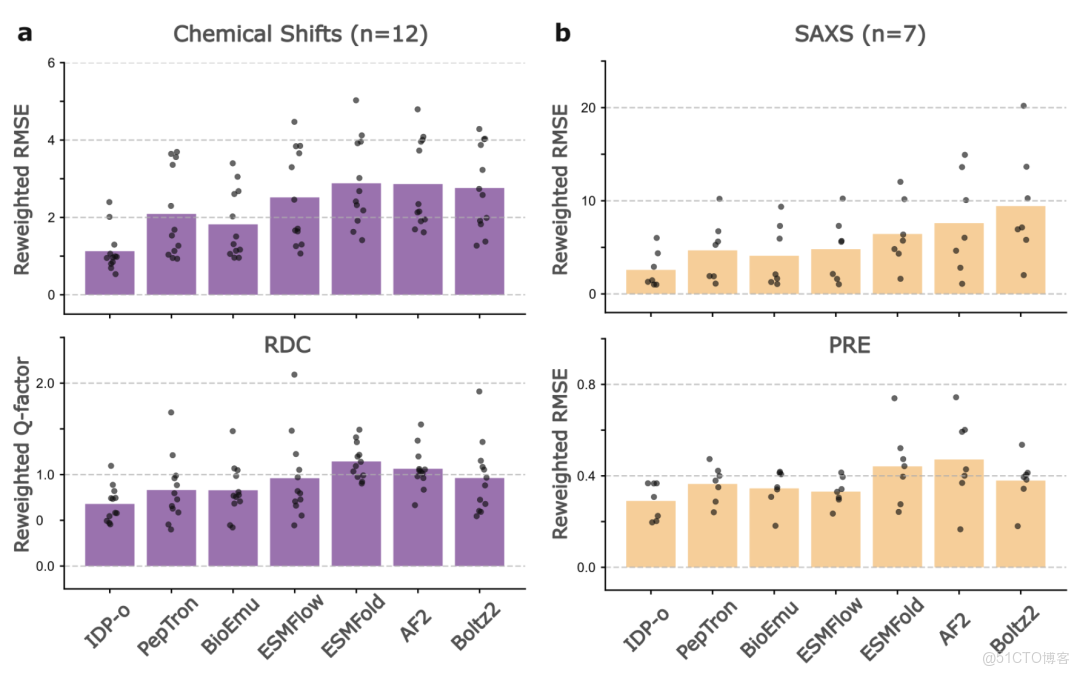
::: hljs-center
不同模型在 PeptoneDB-Integrative 數據集的預測結果
:::
研究也指出了當前模型的幾類共性瓶頸: 多數模型在長程接觸偏好上捕捉不足,存在不同程度的二級結構偏置;此外,對於「條件性摺疊序列」的未摺疊狀態,主流模型普遍難以準確描述,而 IDP-o 在這一方面表現出獨特的優越性。
從無序到有序:IDP 研究的全球突破與產業新篇
內在無序蛋白(IDPs)因其高度動態的構象特徵及與多類重大疾病的緊密相關性,正迅速成為全球生命科學與醫藥產業的研究前沿。
在學術界,AI 結構預測技術正在成為破解 IDP「動態密碼」的關鍵力量。劍橋大學提出的 AlphaFold-Metainference 方法, 將 AlphaFold 的對齊誤差圖與分子動力學模擬相結合,突破了傳統 AlphaFold 主要預測穩定結構的侷限,成功構建了 IDP 及含無序區域蛋白的結構集合,為理解其多態性提供了新路徑。
論文題目:
AlphaFold prediction of structural ensembles of disordered proteins
論文鏈接: https://www.nature.com/articles/s41467-025-56572-9
哥本哈根大學團隊進一步整合 AlphaFold 與蛋白質語言模型,實現了人類無序蛋白質組構象的大規模預測, 驗證了 AI 技術在 IDP 研究中的普適性與可擴展性。
論文題目:
Conformational ensembles of the human intrinsically disordered proteome
論文鏈接: https://www.nature.com/articles/s41586-023-07004-5
而學術成果能否真正改變疾病治療,還取決於產業界的技術轉化能力。英國生物技術公司 Peptone 與德國製藥企業 Evotec 的合作,便展示了 IDP 研究向藥物研發延伸的可行路徑。 依託 Peptone 的超快速氫氘交換質譜(HDX-MS)平台,研究人員可實時追蹤無序蛋白的動態變化,捕捉傳統結構測定手段難以識別的結合位點;再結合 Evotec 在靶點驗證、藥物篩選及臨牀推進方面的優勢,使得難成藥的 IDP 靶點有望轉化為具備成藥潛力的候選分子。
這一系列進展不僅與 PepTron 模型「覆蓋有序—無序結構全譜」的趨勢相互呼應,也標誌着曾被視為難以捉摸的無序蛋白,正逐步成為精準醫學與生物製藥中的關鍵靶點。隨着技術突破與產業協作的不斷加深,IDPs 或將為未來疾病治療提供全新的理解框架與干預路徑。
參考鏈接:
1.https://www.vbdata.cn/intelDetail/717834
2.https://c.m.163.com/news/a/JDIR2LQJ0552ZPM2.html
3.https://www.vbdata.cn/intelDetail/580634